 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
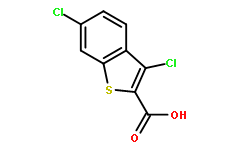




















| Cas No.: | 34576-94-8 |
| Chemical Name: | Benzo[b]thiophene-2-carboxylicacid, 3,6-dichloro- |
| SMILES: | ClC1=CC2=C(C=C1)C(Cl)=C(C(O)=O)S2 |
| Formula: | C9H4O2Scl2 |
| M.Wt: | 247.09786 |
| Purity: | >98% |
| Sotrage: | Powder:-20°C 3 years |
| Publication: | [1]. Tso SC, et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain α-ketoacid dehydrogenase kinase. J Biol Chem. 2014 Jul 25;289(30):20583-93. [2]. Friberg A, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013 Jan 10;56(1):15-30. |
| Description: | BT2 is a BCKDC kinase (BDK) inhibitor with an IC50 of 3.19 μM. BT2 binding to BDK triggers helix movements in the N-terminal domain, resulting in the dissociation of BDK from the branched-chain α-ketoacid dehydrogenase complex (BCKDC)[1]. BT2 (compound 4) is also a potent and selective Mcl-1 inhibitor with a Ki value of 59 μM[2]. |
| Target: | BDK:3.19 μM (IC50) Mcl-1:59 μM (Ki) |
| In Vivo: | BT2 (20 mg/kg/day; intraperitoneal injection; daily; for 7 days; C57BL/6J male mice) treatment robustly enhances BCKDC activity in the heart (12.3-fold) compared with the vehicle-treated animals. Less activation is obtained in muscle and kidney at 3.6- and 3.8-fold, respectively. The -fold activation of BCKDC activity in the above tissues correlates with decreased phosphorylation in heart, muscle, and kidney after the long term BT2 treatment. BT2 treatment reduces the protein levels of BDK in kidneys and heart[1]. Animal Model: C57BL/6J male mice (8-10-week-old)[1] Dosage: 20 mg/kg/day Administration: Intraperitoneal injection; daily; for 1 week Result: BCKDC activity was robustly (12.3-fold) enhanced in the heart compared with the vehicle-treated animals. Less activation was obtained in muscle and kidney at 3.6- and 3.8-fold, respectively. The protein levels of BDK in kidneys and heart were reduced to averages of 39 and 24%, respectively. |
| References: | [1]. Tso SC, et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain α-ketoacid dehydrogenase kinase. J Biol Chem. 2014 Jul 25;289(30):20583-93. [2]. Friberg A, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013 Jan 10;56(1):15-30. |