 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
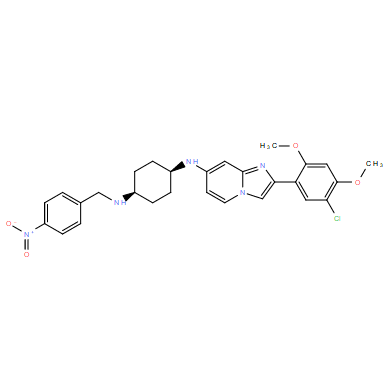
| DC11256 | OTS193320 Featured |
OTS193320 (OTS-193320) is a potent inhibitor of protein methyltransferase SUV39H2 with IC50 of 22.2 nM.
More description
|
|
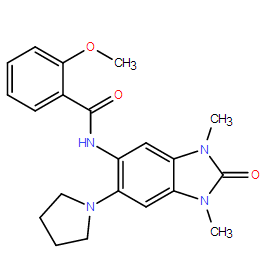
| DC8291 | PFI-4 Featured |
PFI-4 is a potent and selective BRPF1 bromodomain inhibitor (IC50 = 80 nM).
More description
|
|
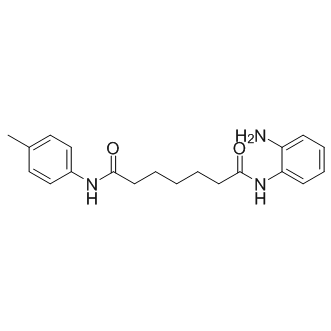
| DC8661 | Pimelic Diphenylamide 106(TC-H 106) Featured |
Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I HDAC (HDAC 1, 2, and 3, with IC50 values of 150 nM , 760nM, and 370 nM, respectively), demonstrating no activity against class II HDACs.
More description
|
|
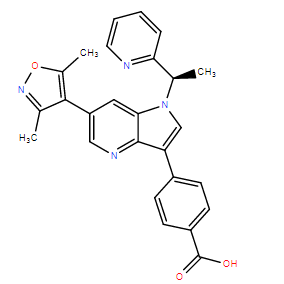
| DC10786 | PLX51107 Featured |
PLX51107 is a novel BET inhibitor with a unique binding mode in the acetylated lysine binding pocket of BRD4 that differentiates it from other compounds under investigation.
More description
|
|
| DC71230 | SDR-04 |
SDR-04 is a BET inhibitor and exhibits strong BRD4-BD1 affinity and inhibition activity. SDR-04 potently suppresses MV4;11 cancer cell line proliferation.
More description
|

|
| DC71179 | GEM144 |
GEM144 is a potent and orally active DNA polymerase α (POLA1) and HDAC 11 dual inhibitor. GEM144 induces acetylation of p53, activation of p21, G1/S cell cycle arrest, and apoptosis. GEM144 has significant antitumor activity in human orthotopic malignant pleural mesothelioma xenografts.
More description
|
|
| DC71108 | RSC133 |
RSC133 exhibits dual activity by inhibiting histone deacetylase and DNA methyltransferase. RSC133 effectively facilitates reprogramming of human somatic cells to pluripotent stem cells and supports the maintenance of an undifferentiated state of human pluripotent stem cells.
More description
|
|
| DC71077 | MAK683 hydrochloride |
MAK683 hydrochloride is an embryonic ectoderm development (EED) inhibitor extracted from patent US20160176882 A1, compound example 2. MAK683 exhibits IC50s of 59, 89, 26 nM in EED Alphascreen binding, LC-MS and ELISA assay.
More description
|
|
| DC71069 | JPS036 |
JPS036 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS036 degrades class I histone deacetylase (HDAC). JPS036 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells.
More description
|
|
| DC71068 | JPS035 |
JPS035 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS035 degrades class I histone deacetylase (HDAC). JPS035 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells.
More description
|
|
| DC71067 | JPS016 |
JPS016 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS016 degrades class I histone deacetylase (HDAC). JPS016 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells.
More description
|
|
| DC71066 | JPS014 |
JPS014 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS014 degrades class I histone deacetylase (HDAC). JPS014 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells.
More description
|
|
| DC71021 | Coumarin-SAHA |
Coumarin-SAHA is a fluorescent probe for determining the binding affinities (kd) and the dissociation off-rates (koff) of the HDAC8-inhibitor complexes.
More description
|
|
| DC70995 | 2-Hexyl-4-pentynoic acid |
2-Hexyl-4-pentynoic acid ((±)-2-Hexyl-4-pentynoic acid), valproic acid (VPA) derivative, exhibits potential roles of HDAC inhibition (IC50=13 µM) and HSP70 induction. Potent neuroprotective effects. 2-Hexyl-4-pentynoic acid causes histone hyperacetylation and protect against glutamate-induced excitotoxicity in cultured neurons.
More description
|
|
| DC70993 | 2′,3′,5′-Triacetyl-5-azacytidine |
2′,3′,5′-Triacetyl-5-azacytidine is an orally active prodrug of 5-Azacytidine. 5-Azacytidine is an inhibitor of DNA methyltransferase.
More description
|
|
| DC70972 | MI-1 |
MI-1 inhibits Menin-MLL interaction with an IC50 of 1.9 μM.
More description
|
|
| DC70945 | [18F]-NT160 |
[18F]-NT160, a Florbetapir (18F)-radiolabeled NT160, is a diagnostic tool for positron emission tomography (PET). NT160 is a brain-penetrant and selective class-IIa HDAC inhibitor with an IC50 of 46 nM. NT160 exhibits a remarkably high inhibition against HDAC4, HDAC5, HDAC7, and HDAC9 with IC50s of 0.08 nM, 1.2 nM, 1.0 nM, and 0.9 nM, respectively.
More description
|
|
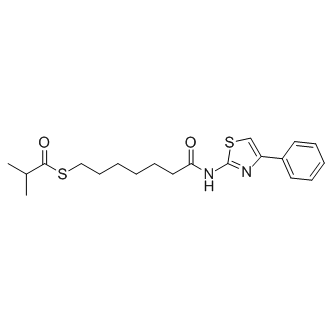
| DC8654 | PTACH (NCH-51) Featured |
PTACH (NCH-51) is a SAHA-based novel inhibitor of human HDAC. PTACH exerts potent growth inhibition against various human cancer cells, with EC50 values ranging from 1 to 10 μM.
More description
|
|
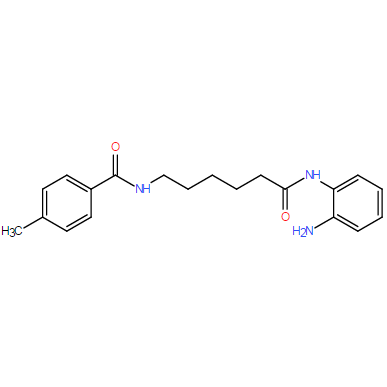
| DC7795 | RG2833 (RGFP109) Featured |
RG2833 is a brain-penetrant inhibitor of HDAC with IC50 values of 60 nM and 50 nM for HDAC1 and HDAC3, respectively.
More description
|
|
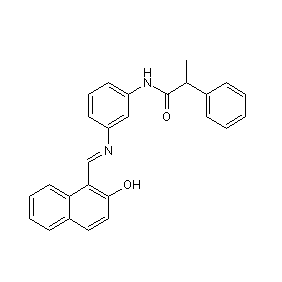
| DC10002 | Salermide Featured |
Salermide is an inhibitor of SIRT1 and SIRT2, causing tumor-specific apoptotic cell death. In MOLT4 leukemia cells, salermide causes 90% apoptosis within 72 hours (IC50 ~ 20 μM) by reactivating proapototic genes that are repressed by SIRT1.
More description
|
|
| DC10429 | SGC2085 Featured |
SGC2085 is a potent and selective coactivator associated arginine methyltransferase 1 (CARM1) inhibitor with an IC50 of 50 nM.
More description
|
|
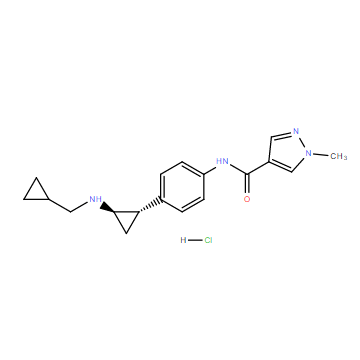
| DC11703 | T-3775440 hydrochloride Featured |
T-3775440 hydrochloride is a novel potent, selecitve, irreversible LSD1 inhibitor with IC50 of 2.1 nM.
More description
|
|
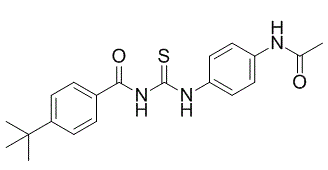
| DC1065 | Tenovin-1 Featured |
Tenovin-1 is a p53 activator and acts through inhibition of protein-deacetylating activities of SirT1 and SirT2.
More description
|
|
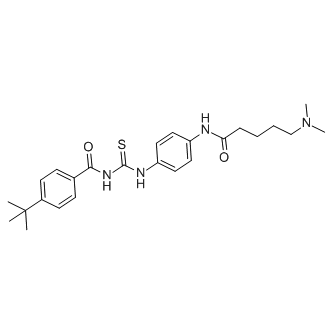
| DC7315 | Tenovin-6 Featured |
Tenovin-6 is the water soluble analog of Tenovin-1 and acts as a potent SIRT1 (IC50=21 uM) and SIRT2 (IC50= 10 uM) inhibitor as well as p53 activator.
More description
|
|
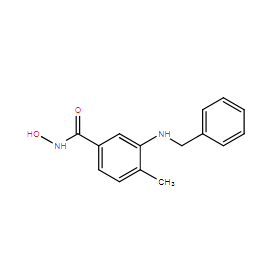
| DC12487 | TH-34 Featured |
TH34 is a potent HDAC6/8/10 inhibitor, induceing DNA damage-mediated cell death in human high-grade neuroblastoma cell lines.
More description
|
|
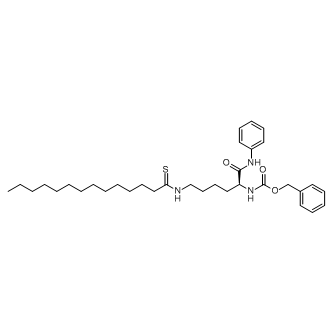
| DC10076 | Thiomyristoyl Featured |
Thiomyristoyl is a potent and specific SIRT2 inhibitor with an IC50 of 28 nM.
More description
|
|
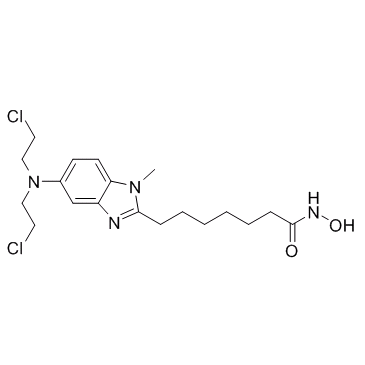
| DC10478 | Tinostamustine(EDO-S101) Featured |
Tinostamustine is the first representative of the A-DAC principle, a new approach in chemotherapy that uses fusion technology to combine an alkylating agent with a pan-histone deacetylase inhibitor (HDAC) to simultaneously damage DNA and block damage repa
More description
|
|
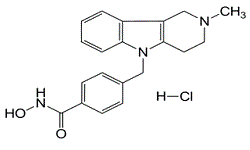
| DC6303 | Tubastatin A HCl Featured |
Tubastatin A is a potent and selective HDAC6 inhibitor with IC50 of 15 nM. It is selective against all the other isozymes (1000-fold) except HDAC8 (57-fold).
More description
|
|
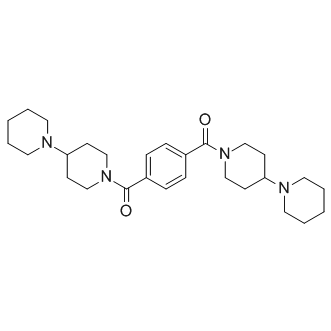
| DC10024 | UNC-1079 Featured |
UNC1079 is the piperidine analog of UNC1021, as a structurally similar but significantly less potent inhibitor for use as a negative control in cellular studies.
More description
|
|
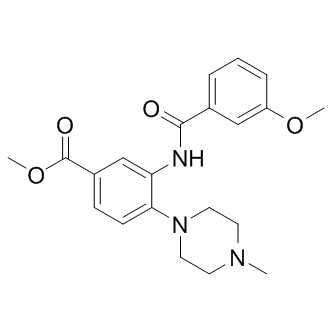
| DC8662 | WDR5-0103 Featured |
WDR5-0103 is a potent and selective WD repeat-containing protein 5 (WDR5) antagonist with Kd of 450 nM.
More description
|
|