 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
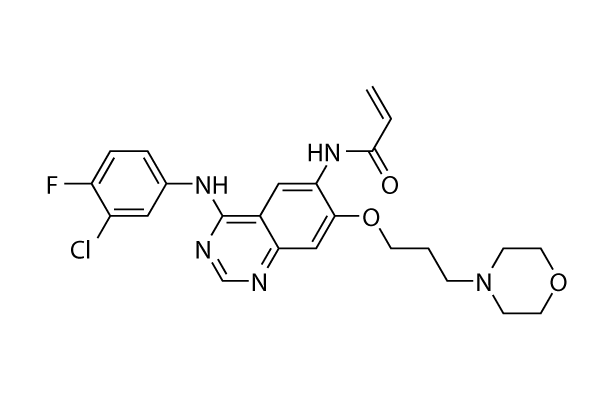




















| Cas No.: | 267243-28-7 |
| Chemical Name: | N-(4-((3-Chloro-4-fluorophenyl)amino)-7-(3-(morpholin-4-yl)propoxy)quinazolin-6-yl)prop-2-enamide |
| Synonyms: | CI1033; CI1033; CI-1033; PD183805; PD183805; PD183805; Canertinib free base; Canertinib |
| SMILES: | C=CC(NC1C=C2C(NC3=CC=C(F)C(Cl)=C3)=NC=NC2=CC=1OCCN1CCOCC1)=O |
| Formula: | C24H25ClFN5O3 |
| M.Wt: | 485.9444 |
| Purity: | >98% |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Canertinib (CI-1033;PD-183805) is a potent and irreversible EGFR inhibitor; inhibits cellular EGFR and ErbB2 autophosphorylation with IC50s of 7.4 and 9 nM. |
| Target: | EGFR:7.4 nM (IC50) ErbB2:9 nM (IC50) |
| In Vivo: | Canertinib shows superior in vivo antitumor activity, giving growth delays in A431 xenografts exceeding 50 days following oral administration[1]. The growth of human malignant melanoma xenografts, RaH3 and RaH5, in nude mice is significantly inhibited by i.p. injections of 40 mg/kg/day canertinib (Fig. 4). The anti-proliferative effect on melanoma xenografts is visible already within 4 days of treatment and further increased throughout the treatment period as observed through the differences in tumor volumes, reaching statistical significance within 18 days of treatment[2]. |
| In Vitro: | Canertinib significantly inhibits growth of cultured melanoma cells, RaH3 and RaH5, in a dose-dependent manner. IC50 is approximately 0.8 μM and by 5μM both cell lines are completely growth-arrested within 72 h of treatment. Incubation of exponentially growing RaH3 and RaH5 with 1 μM canertinib accumulated the cells in the G1-phase of the cell cycle within 24 h of treatment without induction of apoptosis. 1 μM canertinib inhibits ErbB1-3 receptor phosphorylation with a concomitant decrease of Akt-, Erk1/2- and Stat3 activity in both cell lines[2]. |
| Kinase Assay: | Enzyme assays for IC50 determinations are performed in 96-well filter plates. The total volume is 0.1 mL containing 20 mM Hepes, pH 7.4, 50 mM sodium vanadate, 40 mM magnesium chloride, 10 µM adenosine triphosphate (ATP) containing 0.5 mCi of [32P]ATP, 20 mg of polyglutamic acid/tyrosine, 10 ng of EGFR tyrosine kinase, and appropriate dilutions of inhibitor (Canertinib). All components except the ATP are added to the well and the plate is incubated with shaking for 10 min at 25°C. The reaction is started by adding [32P]ATP, and the plate is incubated at 25°C for 10 min. The reaction is terminated by addition of 0.1 mL of 20% trichloroacetic acid (TCA). The plate is kept at 4°C for at least 15 min to allow the substrate to precipitate. The wells is then washed five times with 0.2 mL of 10% TCA and 32P incorporation determined with a plate counter[1]. |
| Cell Assay: | RaH3 and RaH5 cells are treated with increasing concentrations (0-10 μM) of Canertinib for 72 h. The cells are suspended in buffer and counted[2]. |
| Animal Administration: | Mice: Canertinib treatment starts when the tumors show reliable growth. The mice are randomized into control and treatment groups. In the canertinib treated RaH3 group (n=4) and RaH5 group (n=7) each mouse receives i.p. injections of 1.2 mg canertinib (40 mg/kg/day) in 0.1 ml 0.15 M NaCl 5 days a week. The control RaH3 (n=3) and RaH5 (n=7) mice receive i.p. injections of vehicle only according to the same regimen. At the end of the treatment period, the mice are sacrificed by cervical dislocation where after the tumors are removed and weighed[2]. |
| References: | [1]. Smaill JB, et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J Med Chem. 2000 Apr 6;43(7):1380-97. [2]. Djerf Severinsson EA, et al. The pan-ErbB receptor tyrosine kinase inhibitor canertinib promotes apoptosis of malignant melanoma in vitro and displays anti-tumor activity in vivo. Biochem Biophys Res Commun. 2011 Oct 28;414(3):563-8. |