| DC70664 |
NRT-870-59 |
NRT-870-59 is a potent, reversible PTP4A3 inhibitor (IC50=86 nM) with inhibitory specificity for PTP4A3 versus both PTP4A3 A111S mutant and CDC25B.NRT-870-59 does not inhibit CDC25B and PTP4A3 A111S mutant at 1 uM, and 5- to 6-fold less potent aginst DUSP3.NRT-870-59 inhibits PTP4A1, PTP4A2, PTP4A3 mutants C49S, K144I and A106V with IC50 of 133.2, 264.4, 86.0, 332.2, 180.2 and 57.7 nM, respectively.NRT-870-59 inhibits cancer cell colony formation, which is required PTP4A3 expression.The PTP4A (phosphatase of regenerating liver/PRL) phosphatases are a unique subfamily of protein tyrosine phosphatases (PTPs) comprising three highly homologous members (PTP4A1, PTP4A2, and PTP4A3) with 80% amino acid sequence identity.PTP4A3, and to a lesser extent PTP4A1 and PTP4A2, are overexpressed in many types of cancer, promote tumor invasion and dissemination, and contribute to poor patient prognosis. |

|
| DC70683 |
PF-04827736 |
PF-04827736 ((S)-PF-04677490) is a potent, selective PDE1 inhibitor with IC50 of 42, 9.1 and 38 nM for PDE1A, PDE1B and PDE1C, respectively; displays >10-fold selectivity over PDE10A1, and >45-fold selectivity over other PDE isoforms. |
|
| DC70684 |
PF-06427878 |
PF-06427878 is a potent, selective, oral DGAT2 inhibitor with IC50 of 99/202 nM for human/rat DGAT2, >470-fold selectivity over DGAT1 and MGAT1/2/3.PF-06427878 inhibits DGAT2-dependent triglyceride (TG) synthesis in primary human hepatocytes with IC50 of 11.6 nM.PF-06427878 reduces hepatic and circulating plasma TG levels as well as lipogenic gene expression in rats maintained on a Western-type diet (0.3-30 mg/kg bid. po.).PF-06427878 significantly improves steatosis and hepatocellular ballooning with a decrease in lobular inflammation in a murine nonalcoholic steatohepatitis (NASH) model (2 or 20 mg/kg bid. po.). |
|
| DC70691 |
PF-07284892 |
PF-07284892 (PF 07284892) is a small molecule inhibitor of SHP2 that may block MAPK signaling and lead to tumor growth inhibition. |
|
| DC70694 |
PG-116800 |
PG-116800 is a selective, oral matrix metalloproteinase (MMPs) inhibitor with significant antiremodeling effects in animal models of MI and ischemic heart failure. |
|
| DC70700 |
PKM-833 |
PKM-833 (PKM833) is a potent, selective, and orally active FAAH inhibitor with IC50 of 8.8 and 10 nM against human and rat FAAH, respectively.PKM‐833 showed human and rat K inact/K i values with 34 300 ± 10 800 and 128 000 ± 39 800 mol-1 L−1 s-1, respectively.PKM-833 does not inhibit human MAGL and a panel of 137 molecular targets, exhibits weak inhibitory effects (>50%) in five receptor binding assays [PFA, 5‐HT2B, sigma, Na+ channel and Cl‐ channel (GABA gated) at 20 uM.PKM-833 showed excellent brain penetration and good oral bioavailability, and elevated anandamide (AEA) concentrations in the rat brain.PKM-833 significantly attenuated formalin-induced pain responses (3 mg/kg) and improved mechanical allodynia in CFA-induced inflammatory pain (0.3-3 mg/kg) in rat models, without significant side effects on catalepsy and motor coordination up to 30 mg/kg. |
|
| DC70708 |
Prinomastat
Featured
|
Prinomastat (AG3340) is a potent, selective MMP inhibitor with pM affinities for inhibiting gelatinases (MMP-2 and -9, Ki=50-150 pM), MMP-14 and MMP-13; demonstrates broad antitumor activity in a number of tumor models, inhibits glioma invasion or growth of the human malignant glioma cell line U87; also suppresses tumor growth in a malignant glioma tumor model. |
_1.gif)
|
| DC70731 |
Rat C53 peptide |
Rat C53 peptide (C53, pGC-B activator C53) is a novel potent, selective particulate guanylyl cyclase B (pGC-B) activator, activates pGC-B/cGMP signaling pathway, and enhances cGMP generating actions.C53 is highly resistant to NEP degradation and has less interaction with NPRC compared to CNP in vitro.C53 activates the protective pGC-B/cGMP signaling pathway and possesses enhanced cGMP generating actions.C53 inhibits fibroblast proliferation chronically and suppresses the differentiation of fibroblasts to myofibroblasts. |
|
| DC70732 |
Rebimastat |
Rebimastat (BMS-275291, D2163) is a potent, broad spectrum matrix metalloproteinase (MMPs) inhibitor with potential antineoplastic activity, shows nM potency against MMP-1/2/7/9/14.BMS-275291 inhibits tumor growth in a B16BL6 model of experimental metastasis, BMS-275291 treatment results in a dose-dependent reduction in the number of lung metastases.BMS-275291 also inhibits angiogenesis in a murine angiogenesis model with a dose-dependent inhibition of endothelial cell migration.Rebimastat (BMS-275291) induces extracellular matrix degradation, and inhibiting angiogenesis, tumor growth and invasion, and metastasis. |
|
| DC70739 |
RMC-4630
Featured
|
RMC-4630 is a potent and selective inhibitor of SHP2, a central node in the RAS signaling pathway. |
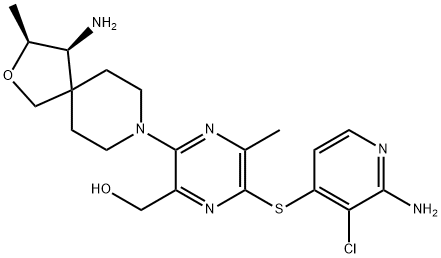
|
| DC70741 |
RO-5459072 |
RO 5459072 (RG-7625) is a potent, highly selective of cathepsin S with IC50 of 0.1 nM and 0.3 nM for human and murine Cathepsin S, respectively; shows no sub-micro molar inhibition against others cathepsins (Cat L, B, K, and F) with the exception of Cat V (IC50=700 nM); dose-dependently reverses aberrant systemic autoimmunity, e.g. plasma cytokines, activation of myeloid cells and hypergammaglobulinemia, especially suppresses IgG autoantibody production. |
|
| DC70758 |
SBI-2130 |
SBI-2130 (SBI 2130) is a novel furanylbenzamide molecule as inhibitor of both WT and oncogenic SHP2 with IC50 of 0.22/5.0 uM for Wt SHP2/SHP2-E76K. |
|
| DC70759 |
SBI-4668 |
SBI-4668 (SBI 4668) is a novel furanylbenzamide molecule as inhibitor of both WT and oncogenic SHP2 with IC50 of 0.73/1.8 uM for Wt SHP2/SHP2-E76K, 13-fold selective over PTP1B and 73-fold selective over STEP;
SBI-4668 exhibited cell viability inhibitory effect on cell growth in both Kasumi-1 cells (IC50=8.5 uM) and KYSE-520 cells (IC50=5.4 uM), SBI-4668 on cell viability was also determined in additional AML cell lines, including MOLM-13 (IC50 = 12 μM) and MV4-11 (IC50 = 8.2 μM).
SBI-4668 inhibited U-937 cell (harbor a G60R oncogenic mutation in SHP2) growth with IC50 of 6.3 uM, which is comparable to the potency found in AML cells expressing WT SHP2, while RMC4550 showed a very weak effect on this G60R mutation cell. |
|
| DC70760 |
SC-78080 |
SC-78080 (SD-2590) is a potent, selective inhibitor of MMP2/9/13 with IC50 of <0.1/0.18/<0.1 nM, respectively.SC-78080 shows weaken inhibitory effect on MMP3/8, and no inhibition on MMP1/7. |
|
| DC70761 |
SCP1 inhibitor T-62 |
SCP1 inhibitor T-62 (T-62) is a specific, small molecule covalent inhibitor of small CTD phosphatase 1 (SCP1), reduces transcription factor REST protein level HEK293 cells. |
|
| DC70762 |
SCP1 inhibitor T-65 |
SCP1 inhibitor T-65 (T-65) is a specific, small molecule covalent inhibitor of small CTD phosphatase 1 (SCP1), reduces transcription factor REST protein level HEK293 cells with EC50 of 1.5 uM.T-65 showed no detectable inhibition against a panel of eight mammalian cysteine-based phosphatases, including SHP2, TCPTP, CD45, DUSP3 and and lymphoid protein tyrosine phosphatase (LYP) at 25 uM.T-65 promotes REST degradation, increases expression of REST-suppressed genes DYRK1a, ELAVL1, USP37, CELSR3, and SCN2 in HEK293 cells. |
|
| DC70787 |
SMIP-30 |
SMIP-30 is a potent and selective inhibitor of PPM1A with IC50 of 1 uM, selectively inhibits the phosphatase activity of PPM1A.SMIP-30 shows poor inhibitory activity against closest homolog PPM1B (IC50>30 uM).SMIP-30 is an uncompetitive inhibitor for PPM1A (Ki=0.84 uM), does not exhibit any cytotoxic effects up to 30 uM in THP-1 macrophages.SMIP-30 dose-dependently reduces the Mtb burden in infected macrophages, and is well tolerated in vivo and combined with rifampicin reduces Mtb burden in the lungs of infected mice.SMIP-30 induces LC3B-II expression, activates autophagy in Mtb-infected macrophages in a PPM1A-dependent manner.SMIP-30 induces a dose-dependent increase in phosphorylation of S403-p62 in WT macrophages. |
|
| DC70806 |
ST-115 |
ST-115 (ST115) is a potent and specific inhibitor of aminopeptidase P2 (XPNPEP2, APP2) with IC50 of 3.7 nM.ST-115 does not inhibit ACE, neprilysin, or multiple other enzymes, and littile inhibition against Aminopeptidase P1/A/B/N (IC50=600 nM->10 uM).ST-115 can reduce bradykinin degradation, increasing its concentration and protective effects when administered intravenously at the start of reperfusion, reduced damaged area of the heart in a mouse model of myocardial infarction.In a rat model of ischemic stroke, ST-115 reduced functional deficits in a skilled walking test by 60% and reduced brain edema by 51%.ST-115 also reduced brain infarct size by 48% in a major subset of rats with small strokes. |
|
| DC70848 |
TK-453 |
TK-453 (TK453) is a potent, selective SHP2 allosteric inhibitor with IC50 of 18.76 and 179 nM for SHP2-WT and SHP2-E76K, respectively.TK-453 has strong inhibitory effect on the enzyme activity of the SHP2-WT, and does not inhibit the activity of the SHP2-PTP catalytic domain, and homologues (SHP1 and PTP1B).TK-453 binds SHP2-WT (KD=150 nM) and SHP2-E76K, higher affinity with SHP2 than SHP099.TK-453 binds in the allosteric binding pocket at the junction of three domains and stabilizes the autoinhibition conformation of SHP2.TK-453 alleviates imiquimod (IMQ)-induced inflammation in macrophages, inhibits the phosphorylation levels of p-IKKα/β and p-p65.TK-453 significantly ameliorates imiquimod-triggered skin-like inflammation in mice via inhibition of IL-23/Th17 axis. |
|
| DC70891 |
VU0285655-1
Featured
|
VU0285655-1 is a potent, selective inhibitor of PLD2, induces autophagy in colorectal cancer cells |
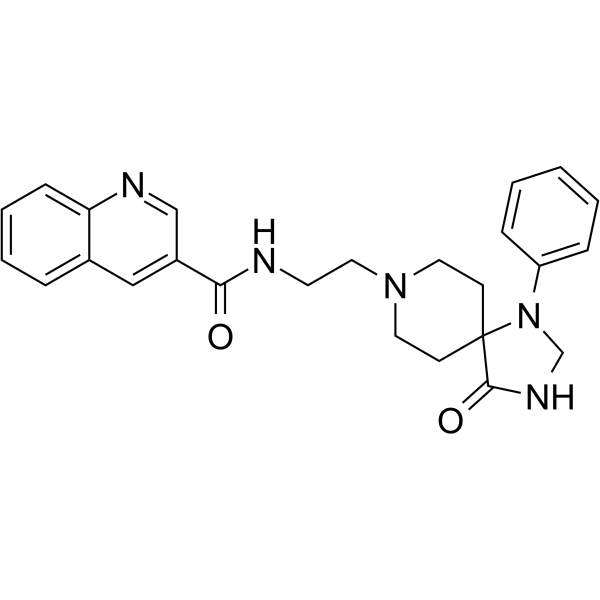
|
| DC70922 |
YTX-7739 |
YTX-7739 is a potent, selective stearoyl-CoA desaturase (SCD) inhibitor with IC50 of 12 nM.YTX-7739 concentration-dependently reduces the fatty acid desaturation index (FADI), the ratio of monounsaturated to saturated fatty acids, with potential to treat synucleinopathies. |
|
| DC70927 |
Z-Arg-Lys-AOMK |
Z-Arg-Lys-AOMK is a potent, selective, irreversible neutral pH 7.2 inhibitor of Cathepsin B with IC50 of 13 nM, >100-fold higher potent than at pH 4.6 (IC50=1,830 nM).
Z-Arg-Lys-AOMK displayed high specificity for cathepsin B compared to other lysosomal cysteine cathepsins.
Z-Arg-Lys-AOMK selectively inhibits cathepsin B cleavage of peptides at neutral cytosolic pH compared to acidic lysosomal pH conditions.
Z-Arg-Lys-AOMK completely inhibited cathepsin B in human neuroblastoma cell lysates at 1 uM.
Z-Arg-Lys-AOMK is cell permeable and inhibits intracellular cathepsin B. |
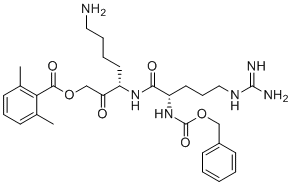
|
| DC70931 |
ZED3197
Featured
|
ZED3197 (ZED-3197) is a novel peptidomimetic direct-acting, potent and selective inhibitor of human FXIIIa, inhibits human plasma derived FXIII‐A2B2 and the recombinant cellular form (FXIII‐A2) with IC50 of 10 and 14 nM, respectively.ZED3197 strongly blocks FXIII‐A2 from mouse, rat, rabbit, dog, and cynomolgus monkey (IC50 8-30 nM), is a significantly weaker inhibitor of FXIII‐A2 from pig (IC50=220 nM).ZED3197 displays 20‐fold to 3000‐fold selectivity against human transglutaminases TG1, TG2, TG3, and TG7.ZED3197 prolonged clot formation, reduced clot firmness, and facilitated clot lysis in whole human blood without affecting the clotting time, indicating minimal impact on hemostasis.ZED3197 effectively decreased the weight of clots and facilitated flow restoration without prolongation of the bleeding time in vivo. |
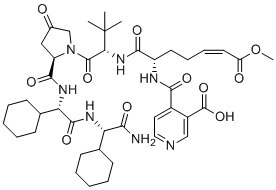
|
| DC70952 |
K203 |
K203 is a potent reactivator of tabun-inhibited AChE. K203 is a crucial antidote used for the organophosphate intoxication. |
|
| DC70988 |
14(S)-HDHA |
14(S)-HDHA (14(S)-HDoHE) is an oxygenation product of Docosahexaenoic acid (DHA). 14(S)-HDHA is a marker reflecting activation of a Docosahexaenoic acid carbon 14-lipoxygenation pathway. |
|
| DC70994 |
2-Fluoropalmitic acid |
2-Fluoropalmitic acid, an acyl-CoA synthetase inhibitor, acts as a candidate anti-glioma agent. 2-Fluoropalmitic acid suppresses the viability and stem-like phenotype of glioma stem cells (GSCs). 2-Fluoropalmitic acid also inhibits proliferation and invasion of glioma cell lines. |
|
| DC71013 |
BOC-L-Phenylalanine-13C |
BOC-L-Phenylalanine-13C is a 13C-labeled BOC-L-Phenylalanine. BOC-L-Phenylalanine is a derivative of Phenylalanine. |
|
| DC71052 |
Harmol |
Harmol categorized as a β-carboline alkaloid. Harmol is a potent MAO inhibitor used as an analytical reference standard. |
|
| DC71055 |
HZ52 |
HZ52 is a potent, reversible 5-lipoxygenase inhibitor, blocking leukotriene synthesis with an IC50 of 0.7 μM in intact human polymorphonuclear leukocytes. |
|
| DC71056 |
Imipramine N-oxide |
Imipramine N-oxide is the metabolite of Imipramine. Imipramine is a tertiary amine tricyclic antidepressant. |
|