- Chemicals for Life Science -
My Cart
0
Home
Services
Process RD
IP Assurance
Out sourcing
Lipid Libraries
Products
Featured products
Novel inhibitors
Natural products
Acids and Aldehydes
Alkaloids
Flavonoids
Other Natural Product
Phenols
Phenylpropanoids
Quinones
Saccharides and Glycosides
Terpenoids
Active Pharmaceutical Ingredient
Therapeutic Antibodies
RNA Delivery
Cationic/Ionizable Lipids
Ionizable Lipids for in vivo CAR
Ionizable Lipids for brain-targeted delivery
Ionizable Lipids for lung-targeted delivery
Classic ionizable lipids for RNA delivery
Clinical-stage ionizable lipids
Ionizable Lipids for solid cancers delivery
Ionizable Lipids for Macrophage delivery
Ionizable Lipids for CRISPR Gene Editing
Ionizable Lipids for circRNA delivery
Ionizable Lipids for muscle delivery
Ionizable Lipids for pancreatic-targeted delivery
Ionizable Lipids for bone marrow-targeted delivery
Ionizable Lipids for lymph-targeted delivery
Ionizable Lipids for eye-targeted delivery
Ionizable Lipids for knee-targeted delivery
Galnac Lipids
Ionizable Lipid library
Classical library-1
Pick Library
LNP kit
PEGylated lipids
Helper lipids
Sterol lipids
Functionalized Lipids
Small molecule tools for RNA delivery
Galnac Molecules
Nucleoside&Phosphoidine Amide
Lipids Custom Synthesis
LNP Formulation Optimization Service
siRNA/ASOs/Aptamer drugs
cap analog
Adjuvant Molecules
TLR agonist small molecule
CpG ODN-TLR9 TLR9 agonist
Adjuvant system
INH-ODN TLR9 Antagonist
Lipids fragment
Lipid Tail
Lipid head group
InVivo Antibodies
InVivo Biosimilar Antibodies
ADC Antibodies
Isotype Controls
Radioligand precursors
Molecular Glues
Inhibitors & Agonists
Antibiotics and Antivirals
Arenavirus
Bacterial
CMV
Enterovirus
Filovirus
Fungal
HBV
HCV
HIV
HPV
HSV
Influenza Virus
Parasite
Prion
Rhinovirus
RSV
SARS-CoV
Virus Protease
Apoptosis
Apaf-1
Apoptosis Inducer
Bcl-2
Caspase
DAPK
Fas Receptor
Ferroptosis
IAP
MDM2-p53
PKD
RIP kinase
TNF-alpha
TRAIL
TNF Receptor
Autophagy
Autophagy-related Protein (Atg)
LRRK2
Mitophagy
Salt Inducible Kinase (SIK)
ULK
WNK Kinase
Cell Cycle/DNA Damage
Antifolate
ATM/ATR
Aurora Kinase
Cdc2-like Kinase (CLK)
Checkpoint Kinase (Chk)
Cyclin-dependent Kinase (CDK)
DNA Alkylator/Crosslinker
DNA Repair Protein
DNA/RNA Synthesis
DNA-PK
Hec1/Nek2
LIM Kinase (LIMK)
Monopolar Spindle 1 (Mps1/TTK)
Nucleoside Antimetabolite/Analog
p21-activated Kinase (PAK)
PARP
PERK
Polo-like Kinase (PLK)
RAD51
ROCK
SRPK
Telomerase
Topoisomerase
Wee1
G-quadruplex
Cytoskeleton/Cell Adhesion Molecules
Arp2/3 complex
Cadherin
Dynamin
Dynein
Ezrin
Galectin
Gap Junction Protein
Integrin
Kinesin
Microtubule/Tubulin
Myosin
Selectin
Epigenetics
Bromodomain
CRISPR/Cas9
DNA Methyltransferase (DNMT)
HDAC
Histone Acetyltransferase (HAT)
Histone Demethylase
Histone Methyltransferase (HMTase)
MicroRNA
Sirtuin
Epigenetic Reader Domain
GPCR
5-HT Receptor
Adenosine Receptor
Adrenergic Receptor
Angiotensin Receptor
Bombesin Receptor
Bradykinin Receptor
Calcium-sensing Receptor (CaSR)
Cannabinoid Receptor
CGRP Receptor
Chemokine Receptor (CCR and CXCR)
Cholecystokinin Receptor
CRF Receptor
Dopamine Receptor
Endothelin Receptor
Free Fatty Acid Receptor (FFAR)
Galanin Receptor
Ghrelin Receptor (GHSR)
Glucagon Receptor
GNRH Receptor
GPCR19 (TGR5)
GPER (GPR30)
GPR109A (Niacin Receptor 1)
GPR119
GPR139
GPR183 (EBI2)
GPR39
GPR55
GPR84
GPR88
Histamine Receptor
Imidazoline Receptor
Leukotriene Receptor
Luteinizing Hormone Receptor (LHR)
Lysophospholipid Receptor
mAChR
Melanin-concentrating Hormone Receptor (MCHR)
Melanocortin Receptor
Melatonin Receptor
mGluR
Motilin Receptor
Neurokinin Receptor
Neuropeptide Y Receptor
Neurotensin Receptor
Opioid Receptor
Orexin Receptor
Oxytocin Receptor (OXTR)
P2Y Receptor
Platelet-activating Factor Receptor
Prostaglandin Receptor
Protease-activated Receptor (PAR)
Proton-sensing GPCRs
PTH Receptor
Relaxin Receptor
Sigma Receptor
Somatostatin Receptor
Taste Receptor
Thyrotropin Receptor
Trace Amine-associated Receptor (TAAR)
Urotensin Receptor
Vasopressin Receptor
GPR24 (MCHR1)
GPR40
Immunology/Inflammation
Complement System
Cyclooxygenase (COX)
FLAP
Interferon Receptor
Interleukin Receptor
IRAK
MyD88
Nitric Oxide Synthase (NOS)
NOD-like Receptor (NLR)
PD-1/PD-L1
PGE synthase
RIG-I-like Receptor (RLR)
Scavenger Receptor
Sphingosine kinase (SphK)
STING
T Cell Receptor (TCR)
Thrombopoietin Receptor (CD110)
Toll-like Receptor (TLR)
Reactive Oxygen Species
JAK/STAT Signaling
JAK
Pim
STAT
Membrane Transporter/Ion Channel
ASBT Transporter (SLC10A2)
BCRP
Calcium Channel
CFTR
Chloride Channel
CRAC Channel
Exportin-1 (CRM1,XPO1)
FABP
GABA Receptor
GABA Transporter (GAT)
Glucose Transporter (GLUT)
Glutamate Transporter
Glycine Transporter (GlyT)
HCN Channel
iGluR
Monoamine Transporter
nAChR
Na-K-ATPase
Na-K-Cl Cotransporter (NKCC)
P2X Receptor
P-glycoprotein (P-gp)
Potassium Channel
Proton Pump
SGLT
Sodium Channel
TRP Channel
URAT1
VDAC
Metabolic Enzyme/Protease
11β-HSD
15-PGDH
Acetylcholinesterase (AChE)
Acetyl-CoA Carboxylase (ACC)
Adenylate Cyclase
Aldehyde Dehydrogenase (ALDH)
Aldose Reductase
Aminopeptidase
Angiotensin-converting Enzyme (ACE)
Arginase
Aromatase
Beta-secretase (BACE)
Calpain
Carbonic Anhydrase
Carboxypeptidase
Cathepsin
COMT
Cytochrome P450 (CYPs)
Diacylglycerol Lipase (DAGL)
Diglyceride Acyltransferase (DGAT)
Dipeptidyl Peptidase (DPP)
Dopamine beta-hydroxylase
Elastase
Enolase
FAAH
Factor Xa
Farnesyl transferase (FTase)
Fatty Acid Synthase
Fumarase
Glucokinase
Glycogen Synthase
Guanylate Cyclase
Gutathione S-transferase (GST)
Hexokinase
HMG-CoA Reductase (HMGCR)
IMPDH
Indoleamine 2,3-Dioxygenase (IDO)
Isocitrate Dehydrogenase (IDH)
Kynurenine 3-Monooxygenase (KMO)
Lactate Dehydrogenase (LDH)
Lipoxygenase
Matrix Metalloproteinase (MMP)
Monoacylglycerol Lipase (MAGL)
Monoamine Oxidase (MAO)
NADPH Oxidase (NOX)
NAMPT
Neprilysin
Phosphodiesterase (PDE)
Phospholipase
Plasminogen Activator Inhibitor-1 (PAI-1)
Protein Arginine Deiminase (PAD)
Protein Phosphatase/PTP
Pyruvate Dehydrogenase Kinase (PDK)
Renin
SGK
S-nitrosoglutathione reductase (GSNOR)
Stearoyl-CoA Desaturase (SCD)
Steroid Sulfatase (STS)
Thrombin
Tryptophan Hydroxylase (TPH)
Xanthine Oxidase (XAO)
Glucosidase
Mitochondrial Metabolism
Drug Metabolite
Endogenous Metabolite
Acyltransferase
Tyrosinase
ATP Citrate Lyase (ACL)
Ser/Thr Protease
NF-κB Pathway
IκB kinase (IKK)
MALT1
NF-κB
NF-κB inducing Kinase (NIK)
TAK1
Nuclear Receptor/Transcription Factor
Androgen Receptor (AR)
Aryl hydrocarbon Receptor (AhR)
c-Fos/AP-1
c-Myc
CREB/CBP
Estrogen Receptor/ERR
Eukaryotic Initiation Factor (eIF)
Farnesoid X Receptor (FXR)
Foxo1
GATA Binding Protein
Glucocorticoid receptor (GR)
HIF/HIF Prolyl-hydroxylase
IRE1
Keap1-Nrf2
Kruppel-like Factor (KLF)
Liver X Receptor (LXR)
Mineralocorticoid Receptor (MLR)
MITF
Nur77 (NR4A1)
NURR1 (NR4A2)
Oct3/4
PGC-1α
PPAR
Pregnane X Receptor (PXR)
Progesterone Receptor
RAR/RXR
Rev-ErbA
ROR
RUNX1-CBFβ
Thyroid Hormone Receptor (THR)
Vitamin D Receptor (VDR)
Yes-associated Protein (YAP)
Others
14-3-3 Protein
Amyloids
AP2-associated Kinase 1 (AAK1)
CaMK
CETP
Choline Kinase (ChoK)
Clathrin
Diacylglycerol Kinase (DGK)
Fluorescent Dye
GRK (GPCRK)
Mitochondrial Permeability Transition Pore (mPTP)
Other Targets
PCSK9
SMN2 Modulator
Spliceosome
Translocator Protein (TSPO)
PI3K/Akt/mTOR
Akt
AMPK
MELK
mTOR
PDK1
PI3K
PI4K
PIKfyve
PTEN
Proteasome/Ubiquitin
Deubiquitinase (DUB)
E3 Ubiquitin Ligase
Heat Shock Protein (HSP)
NEDD8
p97/VCP
Proteasome
Ubiquitin-activating Enzyme (E1)
Ubiquitin-conjugating Enzyme (E2)
Ras-Raf-MAPK-ERK
ERK
JNK
MAP4K
MAPKAPK2 (MK2)
MEK (MAP2K)
MEKK (MAP3K)
Mixed Lineage Kinase (MLK)
MNK
p38 MAPK
Protein Kinase A (PKA)
Raf
Ras
Ribosomal S6 Kinase (RSK)
TOPK
TGF-beta/Smad
PKC
TGF beta Receptor (TGFBR)
Tyrosine Kinase
Ack1 (TNK2)
Anaplastic Lymphoma Kinase (ALK)
Angiopoietin Receptor
Bcr-Abl
BMX Kinase
Breast Tumor Kinase (BRK;PTK6)
BTK
c-Fes Kinase
c-Fms (CSF1R)
c-Kit
c-Met (HGFR)
Discoidin Domain Receptor (DDR)
DYRK
EGFR
Ephrin Receptor
FGFR
FLT3
Focal Adhesion Kinase (FAK)
IGF-1R
Insulin Receptor
ITK
PDGFR
RET Tyrosine Kinase (c-RET)
Src
Syk
TAM Receptor (Tyro3-Axl-Mer)
Tec Kinase
Trk Receptor
VEGFR
Wnt/Notch/Hedgehog
Autotaxin
Axin
Beta-catenin
BMI-1
Casein Kinase
Dickkopf-1 (Dkk-1)
Dishevelled
Frizzled
Gli
GSK-3
Hedgehog (Hh)
Hippo
Notch
Smoothened (Smo)
TNIK
Wnt
γ-secretase
Research Areas
Cancer
Blood Cancer
Bone Cancer
Brain Cancer
Breast Cancer
Chemotherapeutic Agents
Colon Cancer
Gastric Cancer
Kidney Cancer
Liver Cancer
Lung Cancer
Ovarian Cancer
Pancreatic Cancer
Prostate Cancer
Skin Cancer
Solid Tumors
Thyroid Cancer
Infection
Bacterial Infection
CMV Infection
Ebola Virus Infection
Fungal Infection
HBV Infection
HCV Infection
HIV Infection
HPV Infection
HSV Infection
Influenza
Parasite Infection
Rhinovirus Infection
RSV Infection
SARS-CoV Infection
Endocrine Disorders
Cushing Disease
Growth Hormone Deficiency
Hyperparathyroidism
Sexual Dysfunction
Cardiovascular Disease
Atherosclerosis
Coagulation Disorder
Dyslipidemia
Heart Arrhythmia
Heart Failure
Hypercholesterolemia
Hypertension
Ischemia
Thrombosis
Inflammation/Autoimmune Disease
Allergy
Asthma
COPD
Dermatitis
Gout
Multiple Sclerosis
Psoriasis
Rheumatoid Arthritis
Metabolic Disorder
Diabetes
Obesity
Osteoporosis
Neurological Disease
Alcoholism
Alzheimer Disease
Anxiety
Depression
Epilepsy
Migraine
Pain
Parkinson Disease
Schizophrenia
Sleep Disorder
Spinal Muscular Atrophy (SMA)
Stroke
Other Indications
Anemia
Fibrosis
Irritable Bowel Syndrome
Other Indication
Steatohepatitis
Ulcer
Enzymes substrates
PROTACs
E3 Ligase Ligand
E3 Ligase Ligand-Linker Conjugate
PEGn
PROTAC
LYTACs
Antibody Drug Conjugates
ADC Cytotoxin
ADCs Linker/Click Chemistry
Drug-Linker Conjugates
Dyes
Antibody/Protein labeling dyes
How to order
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
Home
>
Inhibitors & Agonists
>
Others
14-3-3 Protein
(1)
Amyloids
(43)
AP2-associated Kinase 1 (AAK1)
(2)
CaMK
(19)
CETP
(9)
Choline Kinase (ChoK)
(1)
Clathrin
(4)
Diacylglycerol Kinase (DGK)
(3)
Fluorescent Dye
(5)
GRK (GPCRK)
(13)
Mitochondrial Permeability Transition Pore (mPTP)
(1)
Other Targets
(16941)
PCSK9
(8)
SMN2 Modulator
(3)
Spliceosome
(10)
Translocator Protein (TSPO)
(4)
Human Antigen R (HuR)
(4)
PHGDH
(3)
Protein Disulfide Isomerase (PDI)
(2)
Pyruvate Kinase
(10)
Regulators of G protein signaling (RGS)
(3)
Others
You can also try the following methods, and our professionals will serve you
Customized Consultation
By Targets:
Rabbit
Mouse
Humanized
Rat
mouse
Chimeric
Cat. No.
Product Name
Field of Application
Chemical Structure
DCX-031
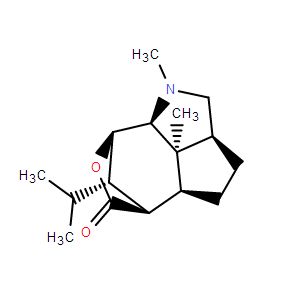
clareolide
>98%,Standard References
More description
DCS-116
Lycodoline
>98%,Standard References
More description
DCS-082
Dendrobine
>98%,Standard References
More description
DCM-008
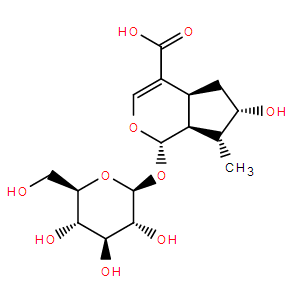
Loganic acid
>98%,Standard References
More description
DCJ-043
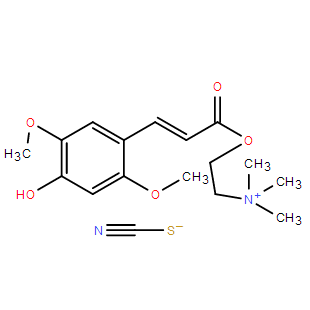
Sinapine thiocyanate
>98%,Standard References
More description
DCY-017
Monocrotaline
>98%,Standard References
More description
DCZ-009
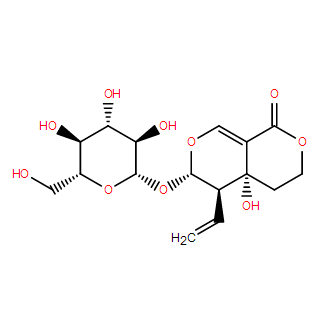
Sweroside
>98%,Standard References
More description
DCS-036
monotropein
>98%,Standard References
More description
DCZ-152
Swertiamarine
>98%,Standard References
More description
DCM-033
toddalolactone
>98%,Standard References
More description
DCS-093
Huperzine B
>98%,Standard References
More description
DCE-034
Dihydrolycorine
>98%,Standard References
More description
DCC-078
Febrifugine
>98%,Standard References
More description
DCY-067
Cryptochlorogenic acid
>98%,Standard References
More description
DCD-062
Scopolin
>98%,Standard References
More description
DCX-014
Neochlorogenic acid
>98%,Standard References
More description
DCS-007
Cimifugin
>98%,Standard References
More description
DCY-118
Lycorine hydrochloride
>98%,Standard References
More description
DCQ-078
Higenamine
>98%,Standard References
More description
DCB-056
Heraclenol
>98%,Standard References
More description
DCY-074
Protosappanin B
>98%,Standard References
More description
DCZ-042
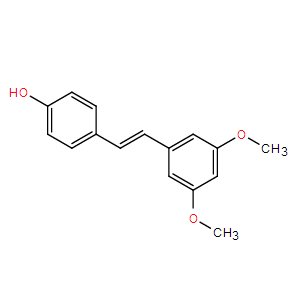
Pterostilbene
>98%,Standard References
More description
DCZ-167
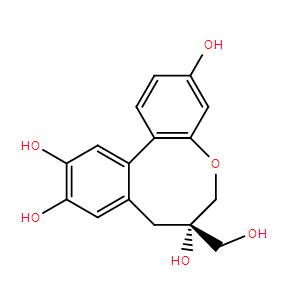
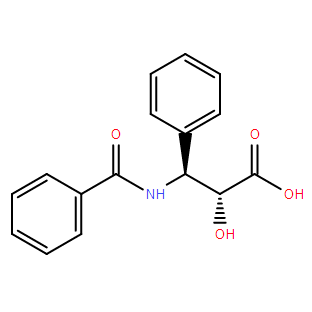
N-Benzoyl-(2R,3S)-3-phenylisoserine
>98%,Standard References
More description
DCC-017
Hesperetin
>98%,Standard References
More description
DCS-088
Hematoxylin
>98%,Standard References
More description
DCD-061
Heraclenin
>98%,Standard References
More description
DCB-060
Brazilin
>98%,Standard References
More description
DCG-034
Licochalcone B
>98%,Standard References
More description
DCY-151
Oxypeucedanin
>98%,Standard References
More description
DCY-007
Isoimperatorin
>98%,Standard References
More description
<
1
...
875
876
877
878
879
...
886
>
GO
10 Article/Page
20 Article/Page
30 Article/Page
Customized Consultation
X
Your information is safe with us. * Required Fields.
Customized Consultation
Your name
Company
Email
Procuct Name
Cat. No.
Remark
Verification code
Please fill out the characters in the picture
X
 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.