 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
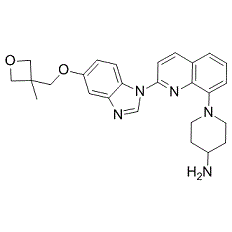




















| Cas No.: | 670220-88-9 |
| Chemical Name: | 1-(2-(5-((3-methyloxetan-3-yl)methoxy)-1H-benzo[d]imidazol-1-yl)quinolin-8-yl)piperidin-4-amine. |
| Synonyms: | CP-868,596; CP-868596; ARO 002;CP868596 |
| SMILES: | NC1CCN(C2=CC=CC3=C2N=C(N4C5=CC=C(C=C5N=C4)OCC6(COC6)C)C=C3)CC1 |
| Formula: | C26H29N5O2 |
| M.Wt: | 443.54 |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 2.1 nM/3.2 nM, 0.74 nM, respectively. |
| In Vivo: | Crenolanib (10 mg/kg and 20 mg/kg) suppresses non-small-cell lung cancer tumor growth in vivo and induces tumor cell apoptosis, and the dosage of crenolanib applied is well tolerated by recipient mice[3]. |
| In Vitro: | Crenolanib has 25-fold more affinity for PDGFRA/B compared with KIT, and is approximately 135-fold more potent than imatinib for inhibiting the PDGFRA D842V mutation. The IC50 for crenolanib for a KIT exon 11 deletion mutant kinase is greater than 1,000 versus 8 nM for imatinib. Crenolanib has low nanomolar potency against the V561D + D842V-mutant kinase that is similar to its potency against the isolated D842V mutation. Both imatinib and crenolanib potently inhibit the kinase activity of the fusion oncogene with IC50 values of 1 and 21 nM, respectively, and inhibits PDGFRA activation in this cell line with IC50 values of 93 and 26 nM, respectively[1]. HL60/VCR and K562/ABCB1 cells, overexpressing ABCB1, are 6.9- and 3.6-fold resistant to crenolanib, respectively, in relation to parental HL60 and K562 cells. PSC-833 fully reverses resistance to crenolanib in both HL60/VCR and K562/ABCB1 cells. Crenolanib (1 nM-10 μM) stimulates ABCB1 ATPase activity in a concentration-dependent manner. Crenolanib treatment does not increase the cell surface expression of ABCB1. Crenolanib inhibits [125I]-IAAP photocrosslinking of ABCB1 at high concentrations, with 50 % inhibition at 10 μM, but has little effect at lower concentrations, below 1 μM[2]. Crenolanib decreases NSCLC cell viability, induces apoptosis in NSCLC cells, and inhibits cell migration in NSCLC cells[3]. |