 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
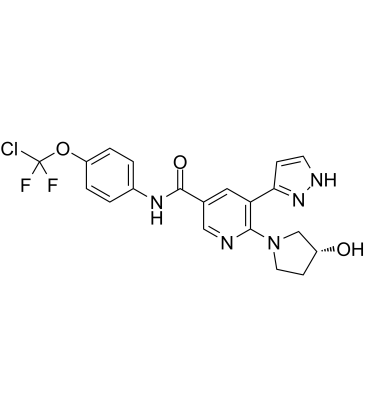




















| Cas No.: | 1492952-76-7 |
| Chemical Name: | Asciminib free base |
| Synonyms: | ABL-001; AB -001; ABL001; asciminib; asciminib free base; |
| SMILES: | O=C(C1=CC(C2=NNC=C2)=C(N3C[C@H](O)CC3)N=C1)NC4=CC=C(OC(F)(Cl)F)C=C4 |
| Formula: | C20H18ClF2N5O3 |
| M.Wt: | 449.843 |
| Purity: | >98% |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Publication: | [1]. Wylie AA, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017 Mar 30;543(7647):733-737. |
| Description: | Asciminib (ABL001) is a potent and selective allosteric Bcr-Abl inhibitor; inhibits Ba/F3 cells grown with an IC50 of 0.25 nM. |
| In Vivo: | Asciminib is undergoing clinical development testing in patients with CML and Philadelphia chromosome-positive acute lymphoblastic leukaemia. Single doses of 7.5, 15 and 30 mg/kg ABL001, administered to mice bearing KCL- 22 xenografts, inhibits pSTAT5 (Tyr694), which return to baseline at 10, 12 and 16-20h after administration of the dose, respectively. In mice implanted with KCL-22 tumors, the minimum dose of asciminib required for complete regression is 7.5 mg/kg twice a day (BID) or 30 mg/kg once a day (QD), and is tolerated at doses up to 250 mg/kg BID. Similarly, in xenografts derived from patients, treatment with 7.5 and 30 mg/kg asciminib leads to regressions that are maintained during dosing[1]. |
| In Vitro: | Asciminib binds to the myristoyl pocket of ABL1 and induces the formation of an inactive kinase conformation. NMR and biophysical studies confirm that asciminib binds potently (dissociation constant=0.5-0.8nM) and selectively to the myristoyl pocket of ABL1 and induces the inactive C-terminal helix conformation. Asciminib binding mimics the structural consequences of myristate binding to the N terminus of ABL1. Consistent with this binding site, asciminib exhibits the same non-ATP-competitive biochemical kinetics as the BCR–ABL inhibitor GNF-2 but with approximately 100-fold greater potency. Asciminib lacks activity against more than 60 kinases, including SRC, and is similarly inactive against G-protein-coupled receptors, ion channels, nuclear receptors and transporters. In BCR–ABL1-transformed Ba/F3 cells grown without IL-3, asciminib has an anti-proliferative with IC50 value of 0.25nM. In the CML blast-phase cell line KCL-22, asciminib inhibits phosphorylation of both STAT5 (Tyr694; pSTAT5) and BCR–ABL1 (Tyr245; pBCR–ABL1) after 1h using concentrations that correlate with those required for inhibition of cell proliferation. Asciminib is selectively active against all BCR–ABL1 lines (IC50 value of 1–20nM), irrespective of the presence of either the p210 or the p190 BCR–ABL1 isoform.[1]. |
| Cell Assay: | Ba/F3 cells are treated with a range concentration of asciminib (0-10000 nM) for 48 h. Cell proliferation is measured using the Britelite luciferase detection assay[1]. |
| Animal Administration: | Mice: Asciminib efficacy in three patient-derived ALL systemic xenograft models (ALL-7015, AL-7119 and AL-7155) is assessed by FACS monitoring of the percentage of CD45+ cells per live cell in blood samples taken at varying time points after dosing with either 7.5 mg/kg BID (group 2) or 30 mg/kg BID (group 3) asciminib for 3 weeks[1]. |
| References: | [1]. Wylie AA, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017 Mar 30;543(7647):733-737. |