 DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
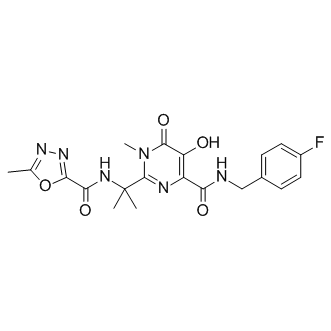




















| Cas No.: | 518048-05-0 |
| Chemical Name: | 4-Pyrimidinecarboxamide, N-((4-fluorophenyl)methyl)-1,6-dihydro-5-hydroxy-1-methyl-2-(1-methyl-1-(((5-methyl-1,3,4-oxadiazol-2-yl)carbonyl)amino)ethyl)-6-oxo- |
| Synonyms: | MK0518; MK 0518; MK-0518; Raltegravir; brand name: Isentress |
| SMILES: | O=C(C(O)=C(C(NCC1=CC=C(C=C1)F)=O)N=C2C(C)(C)NC(C3=NN=C(C)O3)=O)N2C |
| Formula: | C20H21FN6O5 |
| M.Wt: | 444.4234 |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Raltegravir is a potent integrase (IN) inhibitor, used to treat HIV infection. |
| In Vivo: | Raltegravir induces viro-immunological improvement of nonhuman primates with progressing SIVmac251 infection. One non-human primate shows an undetectable viral load following Raltegravir monotherapy[5]. |
| In Vitro: | PFV IN carrying the S217H substitution is 10-fold less susceptible to Raltegravir with IC50 of 900 nM. PFV IN displays 10% of WT activity and is inhibited by Raltegravir with an IC50 of 200 nM, indicating a appr twofold decrease in susceptibility to the IN strand transfer inhibitor (INSTI) compared with WT IN. S217Q PFV IN is as sensitive to Raltegravir as the WT enzyme[1]. Raltegravir is metabolized by glucuronidation, not hepatically. Raltegravir has potent in vitro activity against HIV-1, with a 95% inhibitory concentration of 31±20 nM, in human T lymphoid cell cultures. Raltegravir is also active against HIV-2 when Raltegravir is tested in CEMx174 cells, with an IC95of 6 nM. Raltegravir metabolism occurs primarily through glucuronidation. Drugs that are strong inducers of the glucuronidation enzyme, UGT1A1, significantly reduce Raltegravir concentrations and should not be used. Raltegravir exhibits weak inhibitory effects on hepatic cytochrome P450 activity. Raltegravir does not induce CYP3A4 RNA expression or CYP3A4-dependent testosterone 6-β-hydroxylase activity[2]. Raltegravir cellular permeativity is reduced in the presence of magnesium and calcium[3]. Raltegravir and related HIV-1 integrase (IN) strand transfer inhibitors (INSTIs efficiently block viral replication[4]. In acutely infected human lymphoid CD4+ T-cell lines MT-4 and CEMx174, SIVmac251 replication is efficiently inhibited by Raltegravir, which shows an EC90 in the low nanomolar range[5]. |
MSDS

COA
| LOT NO. | DOWNLOAD |
|
|
|
| 2018-0101 |
|
| 2018-0101 |
|