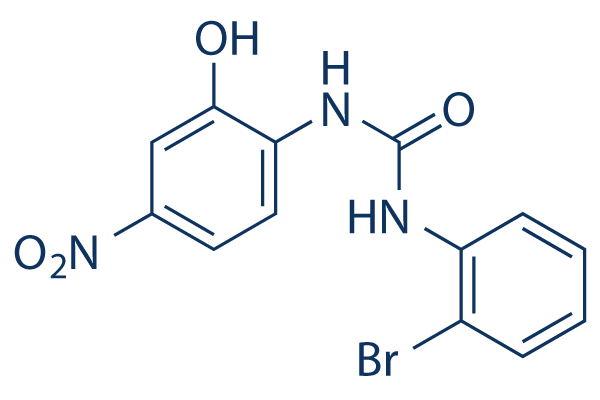
| Description: |
SB225002 is a potent and selective CXCR2 antagonist with an IC50 of 22 nM. |
| Target: |
125I-IL-8-CXCR2:22 nM (IC50, in CHO cell membrane) |
| In Vivo: |
SB225002 (SB 225002) selectively blocks IL-8-induced neutrophil margination in rabbits[1]. CXCR2 is blocked using the selective antagonist SB225002 (2 mg/kg) or neutralizing CXCR2 antiserum. The CXCR2 antagonist SB225002 decreases neutrophil counts in ischemic hemispheres of ApoE−/− mice on Western diet and wildtype mice on normal diet[3]. SB225002 significantly attenuates microglial activation and BBB damage, increases myelination, and reduces astrogliosis in the white matter after LPS-sensitized HI[4]. |
| In Vitro: |
SB225002 (SB 225002) is an antagonist of 125I-IL-8 binding to CXCR2 with an IC50=22 nM. SB225002 shows >150-fold selectivity over CXCR1 and four other 7-TMRs tested. SB225002 is a potent antagonist of rabbit CXCR2, inhibiting rabbit PMN chemotaxis in response to optimal concentrations of human IL-8 or GROα (IC50 values of 30 and 70 nM, respectively. In these cells (PMN, HL60, CXCR1-RBL-2H3), SB225002 produces a concentration-dependent inhibition of both IL-8- and GROα-mediated calcium mobilization with IC50 values of 8 and 10 nM, respectively. In 3ASubE cells stably transfected with CXCR2, SB 225002 dose-dependently inhibits calcium mobilization induced by both GROα and IL-8, with IC50 values of 20 and 40 nM, respectively[1]. WHCO1 cells treated with SB225002 exhibits a 40% reduction in cell proliferation. Blocking CXCR2 signaling in WHCO1 cells with 400 nM SB225002 (SB 225002) significantly decreases cell proliferation by ~40% to 50%[2]. |
| Kinase Assay: |
CHO-CXCR1 and CHO-CXCR2 membranes are prepared. Assays are performed in 96-well microtiter plates where the reaction mixture contained 1.0 μg/mL membrane protein in 20 mM Bis-Tris-propane, pH 8.0, with 1.2 mM MgSO4, 0.1 mM EDTA, 25 mM NaCl, and 0.03% CHAPS and SB 225002 (10 mM stock in Me2SO) added at the indicated concentrations, the final Me2SO concentration is <1% under standard binding conditions. Binding is initiated by addition of 0.25 nM 125I-IL-8 (2,200 Ci/mmol). After 1-h incubation at room temperature the plate is harvested using a Tomtec 96-well harvester onto a glass fiber filtermat blocked with 1% polyethyleneimine, 0.5% BSA and washed three times with 25 mM NaCl, 10 mM Tris•HCl, 1 mM MgSO4, 0.5 mMEDTA, 0.03% CHAPS, pH 7.4. The filter is dried, sealed in a sample bag containing 10 mL of Wallac 205 Betaplate liquid scintillation fluid, and counted with a Wallac 1205 Betaplate liquid scintillation counter[1]. |
| Cell Assay: |
Three esophageal squamous cell carcinoma cell lines WHCO1, WHCO5, and WHCO6 originally established from surgical biopsies of primary esophageal squamous cell carcinomas are cultured in DMEM containing 10% FCS at 37°C in a humidified atmosphere of 5% CO2. MTT assays are carried out using the Cell Proliferation kit. Briefly, 1.5×103 cells are plated in 96-well plates in a final volume of 180 μL DMEM per well. SB 225002 (400 nM) is added to cells and 0.001% DMSO (solvent) is added as a control. After the indicated incubation period, 18 μL of the MTT labeling reagent (final concentration 0.5 mg/mL) is added to each well and incubated for 4 hours in a humidified atmosphere. One hundred eighty microliters of the solubilization solution are added to each well and the plates are left overnight at 37°C. The spectrophotometric absorbance of samples is measured at 595 nm using a microtiter plate reader[2]. |
| Animal Administration: |
Mice[3] Male 7-8 weeks old wildtype (C57BL/6J, Harlan) and ApoE−/− mice, which are generated on the same C57BL/6 background, are either fed with a normal chow or a cholesterol rich chow for 6 weeks and submitted to 20 min of left-sided middle cerebral artery occlusion (MCAO) or sham surgery. Animals are randomly attributed to treatment paradigms, and experimenters are blinded at all stages of interventions and data analysis. The selective CXCR2 antagonist SB225002 (2 mg/kg) or vehicle (1% DMSO in PBS) is injected intraperitoneally (i.p.) at 0, 24 and 48 hours post-ischemia. In other experiments, CXCR2 is specifically blocked by i.p. injection of a neutralizing rabbit anti-CXCR2 serum (300 μL) at 0 hours, 24 hours and 48 hours post-ischemia. In the latter studies, normal rabbit serum (NRS) served as control. In some experiments, neutrophils are depleted by i.p. injection of 200 μg anti-mouse Ly6G 24 hours before and 24 hours after ischemia. In these experiments, 200 μg of an isotype control antibody is delivered as control. Rats[4] In this study, 10-12 Sprague-Dawley rat pups per dam are used. The pups receive intraperitoneal injections of SB225002 (1 or 3 mg/kg, diluted in NS containing 0.33 % Tween 80) or vehicle (NS solution containing 0.33 % Tween 80) 30 min before lipopolysaccharide (LPS) administration and immediately after hypoxic ischemia (HI). The pups are randomly assigned to four groups: control (pups unexposed to LPS or HI, N=14), vehicle (NS injections 30 min before LPS administration and immediately after HI, N=18), and SB-1 (1 mg/kg, N=14) and SB-3 (3 mg/kg, N=18) (SB225002 injections 30 min before LPS administration and immediately after HI). |
| References: |
[1]. White JR, et al. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem. 1998 Apr 24;273(17):10095-8.
[2]. Wang B, et al. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006 Mar 15;66(6):3071-7.
[3]. Herz J, et al. Role of Neutrophils in Exacerbation of Brain Injury After Focal Cerebral Ischemia in Hyperlipidemic Mice. Stroke. 2015 Oct;46(10):2916-25.
[4]. Wang LY, et al. CXCL5 signaling is a shared pathway of neuroinflammation and blood-brain barrier injury contributing to white matter injury in the immature brain. J Neuroinflammation. 2016 Jan 6;13:6.
[5]. Shi ZR, et al. Decrease of galectin-3 in keratinocytes: A potential diagnostic marker and a critical contributor to the pathogenesis of psoriasis. J Autoimmun. 2018 May;89:30-40. |
 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.