 DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
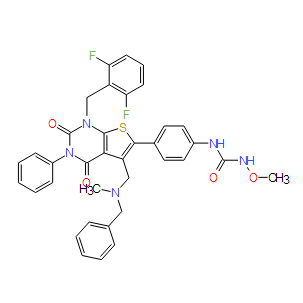




















| Cas No.: | 308831-61-0 |
| Chemical Name: | 1-[4-[5-[[benzyl(methyl)amino]methyl]-1-[(2,6-difluorophenyl)methyl]-2,4-dioxo-3-phenylthieno[2,3-d]pyrimidin-6-yl]phenyl]-3-methoxyurea |
| Synonyms: | TAK013,TAK 013 |
| SMILES: | CN(CC1=CC=CC=C1)CC2=C(SC3=C2C(=O)N(C(=O)N3CC4=C(C=CC=C4F)F)C5=CC=CC=C5)C6=CC=C(C=C6)NC(=O)NOC |
| Formula: | C36H31F2N5O4S |
| M.Wt: | 667.724 |
| Purity: | >98% |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Sufugolix (TAK-013) is a highly potent and orally available luteinizing hormone-releasing hormone (LHRH) receptor antagonist with an IC50 of 0.1 nM. |
| Target: | IC50: 0.1 nM (human LHRH), 0.6 nM (monkey LHRH)[1] |
| In Vivo: | Oral administration of sufugolix causes almost complete suppression of the plasma LH levels in castrated male cynomolgus monkeys at a 30 mg/kg dose with sufficient duration of action (more than 24 h). The maximum plasma concentrations of sufugolix are 0.34 μM (reached 6 h after administration) and 0.18 μM (reached 4 h after administration) at 30 and 10 mg/kg doses, respectively[1]. |
| In Vitro: | Sufugolix exhibits more than 3- and 2000-fold selectivity for the human receptor over the monkey and rat receptors, respectively. Sufugolix effectively antagonizes LHRH function on CHO cells expressing the human (IC50=0.1 nM) and monkey (IC50=0.6 nM) receptors. During the conformational analysis of sufugolix, using high-temperature molecular dynamics calculation, it is observed that the cis conformer of the methoxyurea is more populated than the trans conformer [1]. |
| Kinase Assay: | The receptor-expressing CHO cells are seeded into 24-well plates at a density of 4×104 cells/well and cultured for 1 day. The cells are then incubated with [5,6,8,9,11,12,14,15-3H]arachidonic acid (11 kBq/well) for 1 day and ished with DMEM supplemented with 20 mM HEPES and 0.2% BSA. The cells are then preincubated with the compounds (Sufugolix) at 37 °C for 60 min and the reaction is started by addition of LHRH (1 nM). After incubation at37 °C for 40 min, radioactivity in the medium is measured with a liquid scintillation counter[1]. |
| Animal Administration: | Monkeys: Sufugolix (10 or 30 mg/kg, 3 mL/kg, n=3 for each group) is suspended in 0.5% methylcellulose containing 1.2% citric acid, or 0.5% methylcellulose containing 1.2% citric acid alone (3 mL/kg, n=3), are administered orally. Blood samples (heparin-plasma) are collected from a femoral vein 24 h before administration and 0, 2, 4, 8, 24, and 48 h after administration. LH concentrations in the plasma are measured by bioassays using mouse testicular cells[1]. |
| References: | [1]. Sasaki S, et al. Discovery of a thieno[2,3-d]pyrimidine-2,4-dione bearing a p-methoxyureidophenyl moiety at the 6-position: a highly potent and orally bioavailable non-peptide antagonist for the human luteinizing hormone-releasing hormone receptor. J Med Chem. 2003 Jan 2;46(1):113-24. |
MSDS

COA
| LOT NO. | DOWNLOAD |
|
|
|
| 2018-0101 |
|
| 2018-0101 |
|
| 2018-0101 |
|