| Description: |
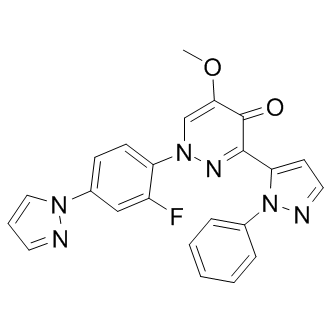
Balipodect (TAK-063) is a highly potent, selective and orally active PDE10A inhibitor with IC50 of 0.30 nM; >15000-fold selectivity over other PDEs. |
| Target: |
IC50: 0.3 nM (PDE10A)[1]. |
| In Vivo: |
Balipodect (TAK-063) has excellent selectivity (>15000-fold selectivity over other PDEs), and favorable pharmacokinetics, including high brain penetration, in mice. Oral administration of Balipodect (TAK-063) to mice elevated striatal 3',5'-cyclic adenosine monophosphate (cAMP) and 3',5'-cyclic guanosine monophosphate (cGMP) levels at 0.3 mg/kg and showed potent suppression of phencyclidine (PCP)-induced hyperlocomotion at a minimum effective dose (MED) of 0.3 mg/kg[1]. Balipodect (TAK-063) at 0.3 and 1 mg/kg, p.o., increased cAMP and cGMP levels in the rodent striatum and upregulated phosphorylation levels of key substrates of cAMP-dependent and cGMP-dependent protein kinases. Balipodect (TAK-063) at 0.3 and 1 mg/kg, p.o., strongly suppressed MK-801-induced hyperlocomotion that is often used as a predictive model for antipsychotic-like activity in rodents. Balipodect (TAK-063) did not affect plasma prolactin or glucose levels at doses up to 3 mg/kg, p.o. Balipodect (TAK-063) at 3 mg/kg, p.o., elicited a weak cataleptic response compared with haloperidol and olanzapine[2]. |
| Animal Administration: |
Mice[1] Compound 27h was suspended in 0.5% (w/v) methylcellulose in distilled water. Compound 27h was administered orally (po). PCP hydrochloride (lot no. 010M4010) purchased from Sigma-Aldrich, Inc. (USA) was dissolved in saline and was administered subcutaneously (sc). All compounds were dosed in a volume of 20 mL/kg body weight. Hyperlocomotion was measured using a spontaneous motor analyzer MDC system (BrainScience idea. Co. Ltd., Japan). Mice were placed in locomotor chambers for more than 60 min for habituation. Animals were removed from each chamber and treated with either vehicle or compound 27h and then quickly returned to the chamber. The following doses were used: 0.1, 0.3, and 1.0 mg/kg, po. Sixty minutes after treatment with compound 27h, animals were again removed from the chambers and treated with either vehicle (saline) or PCP (5 mg/kg as a salt, sc) and then quickly transferred to the test chamber. Activity counts were recorded in successive 1 min bins, and the total number counts were determined for the 120 min period after PCP administration. |
| References: |
[1]. Kunitomo J, et al. Discovery of 1-[2-Fluoro-4-(1H-pyrazol-1-yl)phenyl]-5-methoxy-3-(1-phenyl-1H-pyrazol-5-yl)pyridazin-4(1H)-one (TAK-063), a Highly Potent, Selective, and Orally Active Phosphodiesterase 10A (PDE10A) Inhibitor. J Med Chem. 2014 Nov 26;57(22):9627-43.
[2]. Suzuki K, et al. In Vivo Pharmacological Characterization of TAK-063, a Potent and Selective Phosphodiesterase 10A Inhibitor with Antipsychotic-Like Activity in Rodents. J Pharmacol Exp Ther. 2014 Dec 18. pii: jpet.114.218552. |