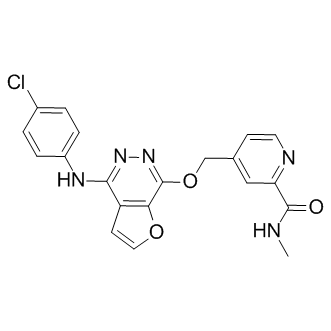
| Description: |
Telatinib (Bay 57-9352) is an orally active, small molecule inhibitor of VEGFR2, VEGFR3, PDGFα, and c-Kit with IC50s of 6, 4, 15 and 1 nM, respectively. |
| Target: |
VEGFR2:6 nM (IC50)
VEGFR3:4 nM (IC50)
PDGFRα:15 nM (IC50)
c-Kit:1 nM (IC50) |
| In Vivo: |
Telatinib causes a significant decrease in endothelium-dependent and endothelium-independent vasodilation. VEGF inhibition by itself decreases NO synthesis, which promotes vasoconstriction, increases peripheral resistance, and therefore can induce an increase in blood pressure[1]. Telatinib (15 mg/kg) with doxorubicin (1.8 mg/kg) significantly decreases the growth rate and tumor size of ABCG2 overexpressing tumors in a xenograft nude mouse model[4]. |
| In Vitro: |
Telatinib has low affinity for the Raf kinase pathway, epidermal growth factor receptor family, the fibroblast growth factor receptor (FGFR) family, or the Tie-2 receptor[2]. Telatinib is metabolized by various cytochrome P450 (CYP) isoforms including CYP3A4/3A5, CYP2C8, CYP2C9, and CYP2C19 as well as by uridine diphosphate glucuronosyltransferase 1A4 (UGT1A4), with the formation of the N-glucuronides of telatinib as the major biotransformation pathway in man. In vitro studies show telatinib to be a weak substrate of the adenosine triphosphate binding cassette (ABC) B1 (ABCB1) transporter[3]. Telatinib at 1 μM significantly enhances the intracellular accumulation of [3H]-mitoxantrone (MX) in ABCG2-overexpressing cell lines. In addition, telatinib at 1 μM significantly reduces the rate of [3H]-MX efflux from ABCG2-overexpressing cells. Furthermore, telatinib significantly inhibits ABCG2-mediated transport of [3H]-E217βG in ABCG2 overexpressing membrane vesicles[4]. |
| Kinase Assay: |
The vanadate (Vi)-sensitive ATPase activity of ABCG2 in the membrane of High Five insect cells is measured. Briefly, membrane (2 μg/0.06 mL) are incubated in ATPase assay buffer with or without 0.4 mM vanadate at 37°C for 5 min and then incubated with varying concentrations of telatinib at 37°C for 5 min. The ATPase reaction is started by the addition of 4 mM Mg-ATP. After incubating at 37°C for 10 min, the reactions are stopped by adding 0.05 mL of 10% SDS solution. The liberated inorganic phosphate is measured[4]. |
| Animal Administration: |
Mice: The mice are randomized into four groups and treated with one of the following regimens: (a) vehicle (10% N-methyl-pyrrolidinone, 90% polyethylene glycol 300) (q3d×6), (b) DOX (1.8 mg/kg, i.p., q3d×6), (c) telatinib dissolved in 10% N-methyl-pyrrolidinone, 90% polyethylene glycol 300 (15 mg/kg, p.o., every 2nd and 3rd day; total 12 times), and (d) DOX (1.8 mg/kg, i.p., q3d×6) + telatinib (15 mg/kg, p.o., every 2nd and 3rd day, given 1 h before giving DOX; total 12 times). DOX for injection is prepared by dissolving in saline. Tumor volume is measured using calipers and body weights are recorded[4]. |
| References: |
[1]. Steeghs N, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. 2008 Jun 1;14(11):3470-6.
[2]. Langenberg MH, et al. Phase I evaluation of telatinib, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in combination with irinotecan and capecitabine in patients with advanced solid tumors. Clin Cancer Res. 2010 Apr 1;16(7):2187-97.
[3]. Steeghs N, et al. Pharmacogenetics of telatinib, a VEGFR-2 and VEGFR-3 tyrosine kinase inhibitor, used in patients with solid tumors. Invest New Drugs. 2011 Feb;29(1):137-43.
[4]. Sodani K, et al. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem Pharmacol. 2014 May 1;89(1):52-61. |