| DC49566 |
Corydamine |
Corydamine, 3-arylisoquinoline alkaloid, is a potent DNA topoisomerase I/II inhibitor. Corydamine has anti-cancer activity. |

|
| DC49567 |
Topoisomerase I/II inhibitor 2 |
Topoisomerase I/II inhibitor 2 (compound 1a) is a potent Topoisomerase inhibitor (IC50= 9.82 μM on Huh7 cells and 6.83 μM on LM9 cells). Topoisomerase I/II inhibitor 2 has dual inhibition on DNA topoisomerase I/II, also can obviously reduce the growth of xenograft tumor in mice model. Topoisomerase I/II inhibitor 2 has the potential value in treating liver cancer. |
|
| DC49568 |
MM41 |
MM41 is a potent stabilizer of human telomeric and gene promoter DNA quadruplexes. MM41 is potently against the MIA PaCa-2 pancreatic cancer cell line with IC50 value of <10 nM. |
|
| DC49569 |
L5-DA |
L5-DA is a G-quadruplex (G4) ligand and selectively stabilized for G4s over ds26. L5-DA exhibits significant cytotoxicity against HeLa cells (IC50=4.3 μM). L5-DA stabilizes G4s in HeLa cells, induces apoptosis, and cell cycle arrest. |
|
| DC50174 |
(E/Z)-Zotiraciclib hydrochloride |
(E/Z)-Zotiraciclib ((E/Z)-TG02) hydrochloride is a potent CDK2, JAK2, and FLT3 inhibitor. |
|
| DC50175 |
CDK4/6-IN-11 |
CDK4/6-IN-11 is a potent PROTAC CDK4/6 degrader. |
|
| DC50176 |
(R)-CTX-712 |
(R)-CTX-712 is a potent inhibitor of cdc2-like kinase (CLK). (R)-CTX-712 inhibits CLK kinase activity, and thus inhibits cancer survival and cancer cell growth. (R)-CTX-712 has the potential for the research of cancer disease (extracted from patent JPWO2017188374A1, compound 286). |
|
| DC50177 |
CDK7-IN-2 |
CDK7-IN-2 is a potent inhibitor of CDK7. CDK7 is implicated in both temporal control of the cell cycle and transcriptional activity. CDK7 is implicated in the transcriptional initiation process by phosphorylation of Rbpl subunit of RNA Polymerase II (RNAPII). CDK7 has the potential for the research of cancer disease, in particular aggressive and hard- to-treat cancers (extracted from patent WO2019099298A1, compound 1). |
|
| DC50178 |
CDK5-IN-3 |
CDK5-IN-3 (compound 11) is a potent and selective CDK5 inhibitor, with IC50s of 0.6 nM and 18 nM for CDK5/p25 and CDK2/CycA, respectively. CDK5-IN-3 can be used for the research of autosomal dominant polycystic kidney disease (ADPKD). |
|
| DC50179 |
SZ-015268 |
SZ-015268 is a CDK7 inhibitor with an IC50 of 23.56 nM. SZ-015268 has extremely significant anti-tumor advantages. SZ-015268 inhibits HCC70, OVCAR-3, HCT116 and HCC1806 cells proliferation with IC50s of 33, 80.56, 12.53, and 61.55 nM, respectively. |
|
| DC50180 |
CDK7-IN-12 |
CDK7-IN-12 is a potent inhibitor of CDK7. CDK7-IN-12 plays a key role in transcriptional regulation and cell cycle regulation. CDK7-IN-12 effectively inhibit malignant tumor proliferation in vitro and in vivo. CDK7-IN-12 has the potential for the research of cancer disease (extracted from patent WO2021249417A1, compound 43). |
|
| DC50181 |
CDK1-IN-1 |
CDK1-IN-7 is a potent CDK1 inhibitor (CDK1/CycB IC50=161.2 nM) with potential antiproliferative activity and selectivity for cancer tissues. CDK1-IN-7 induces apoptosis in p53 dependent manner through the intrinsic apoptotic pathway. CDK1-IN-7 is a potential targeted antitumor agent. |
|
| DC50182 |
FLT3/CDK4-IN-1 |
FLT3/CDK4-IN-1 is a potent, high selective and orally active FLT3/CDK4 dual inhibitor (IC50=11 and 7 nM for FLT3 and CDK4, respectively). FLT3/CDK4-IN-1 has antiproliferative activities against certain cancer cells. FLT3/CDK4-IN-1 has good antitumor effect in vivo. |
|
| DC50185 |
NSC693868 |
NSC693868 is a selective inhibitor of CDK1 and CDK5 with IC50s of 600 nM and 400 nM, respectively. NSC693868 less potently inhibits GSK3β with an IC50 of 1 µM) and does not block CDC25 activity. NSC693868 is used to help define the roles of CDK1 and CDK5 in various signaling pathways. |
|
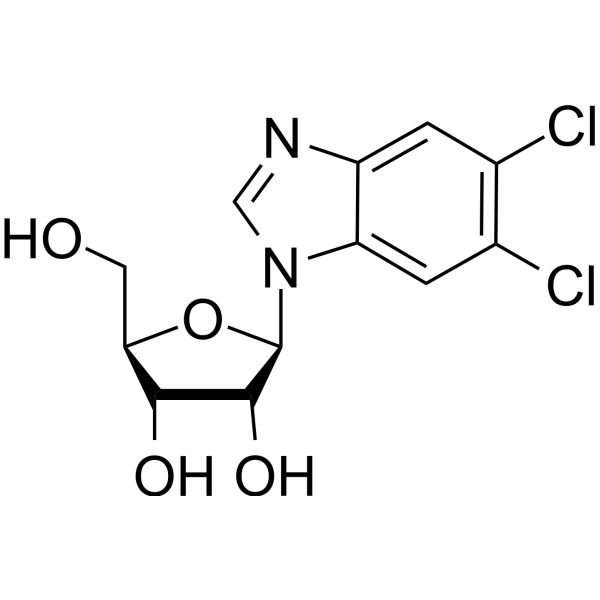
| DC50186 |
5,6-Dichlorobenzimidazole riboside(DBR)
Featured
|
5,6-Dichlorobenzimidazole riboside is a nucleoside analog that inhibits several carboxyl-terminal domain (CTD) kinases including casein kinase II and CDKs. 5,6-Dichlorobenzimidazole riboside trigger p53-dependent apoptosis of human colon adenocarcinoma cells without inducing genotoxic stress to healthy cells. |
|
| DC50188 |
(E/Z)-Zotiraciclib citrate |
(E/Z)-Zotiraciclib citrate is a potent CDK2, JAK2, and FLT3 inhibitor. |
|
| DC50189 |
Anticancer agent 30 |
Anticancer agent 30 (compound 6f-Z), a 3-arylidene-2-oxindole derivative, is a selective CDK2 inhibitor with potent anticancer activity. |
|
| DC50191 |
Anticancer agent 29 |
Anticancer agent 29 (Compd E/Z-6f) is an anticancer agent, with IC50 values of 0.054 μM, 0.127 μM, 0.129 μM, 0.396 μM for CDK2, CDK1, CDK4 and CDK6, respectively. |
|
| DC50192 |
DD-03-156 |
DD-03-156 is a potent and selective degrader of CDK17 and LIMK2. The selectivity and potency of DD-03-156 is exquisite and makes an advanced starting point for the development of a chemical probe for the degradation of CDK17. |
|
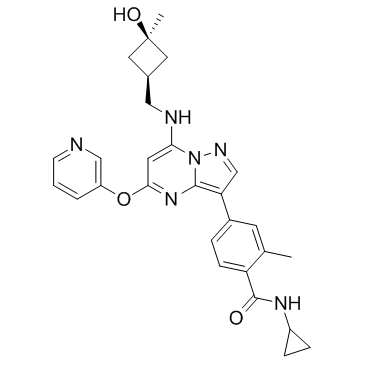
| DC70063 |
CFI-402257(luvixasertib)
Featured
|
CFI-402257(luvixasertib) is a potent and selective Mps1/TTK kinase inhibitor with Ki of 0.1 nM and cellular Mps1 EC50 of 6.5 nM. |
|
| DC70071 |
THZ-1 hydrochloride |
THZ-1 hydrochloride is a potent, selective, covalent CDK7 inhibitor with IC50 of 3.2 nM. |
|
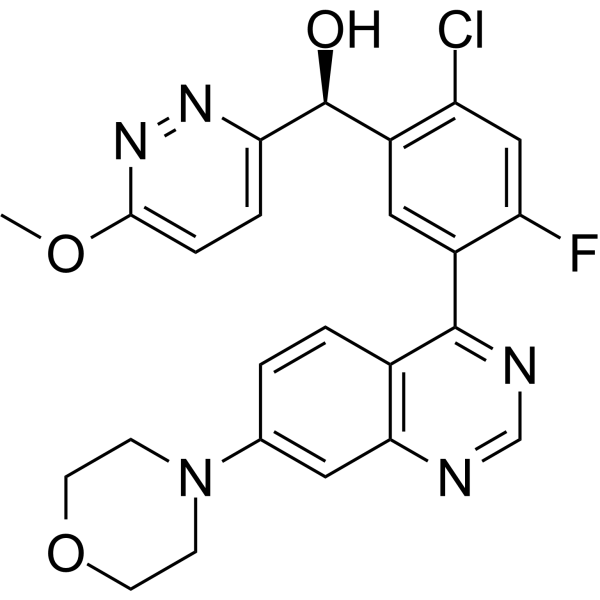
| DC70085 |
Nedisertib (Synonyms: Peposertib; M3814)
Featured
|
Nedisertib (M3814, MSC2490484A) is a highly potent, selective, orally bioavailable inhibitor of DNA-PK. |
|
| DC70116 |
3-Deazauridine |
3-Deazauridine (DAU, NSC 126849) is a nucleoside analog that competitively inhibits cytidine triphosphate synthetase (CTP).3-Deazauridine inhibits the growth of L1210 leukemia cells when used at a concentration of 6 µM and dose-dependently reduces mortality in a mouse model of leukemia.3-Deazauridine selectively suppresses cell viability in a MYC-dependent manner in ARPE-19 cells.3-Deazauridine causes selective replication stress in MYC-overexpressing cells, which originates from MYC-driven rRNA synthesis.3-Deazauridine combined with ATR inhibitor BAY-1895344 induces synthetic lethality to MYC-overexpressing cells, and suppresses tumor growth in 3D assays and in vivo. |
|
| DC70137 |
Nek2 inhibitor 11 |
A potent, ATP-competitive and relative selective Nek2 inhibitor with IC50 of 0.67 uM; displays >10-fold selectivity over CDK2. |
|
| DC70138 |
Nek2 inhibitor 3a |
A potent, relatively selective Nek2 inhibitor with IC50 of 82.74 nM in cell-free assays; inhibits only 3 kinases (YSK4, FLT3-ITDD835V and FLT3-ITDF691L) against 97 kinases with >65% inhibition at 15 nM; blocks NEK2-EZH2 complex formation and inhibits tumor cell proliferation; efficiently attenuates GBM growth in mouse model and exhibits a synergistic effect with radiotherapy. |
|
| DC70157 |
A-1592668 |
A-1592668 (A 1592668) is a novel potent, CDK9-selective inhibitor with IC50 of 1.2 nM, >1000 fold selectivity over CDK1/2/7/8;
A-1592668 also displays selectivity of ~12- and 240-fold over CDK4 and CDK6 respectively.
A-1467729 inhibited phosphorylation of RNA pol-II (p-RNA pol-II; Ser2) with a potency of 26.7 nM at the cellular level, inhibited MCL-1 dependent hematologic tumor cell lines H929 (IC50=39 nM) and MV4-11 (IC50=42.4 nM) following 24 h of incubation. |
|
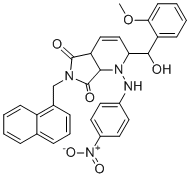
| DC70162 |
A83B4C63
Featured
|
A83B4C63 is a highly potent small-molecule inhibitor targeting the DNA repair enzyme polynucleotide kinase 3'-phosphatase (PNKP), exhibiting strong binding affinity with a Kd of 80 nM. Due to its limited aqueous solubility, A83B4C63 was formulated into nano-encapsulated carriers, enhancing its therapeutic potential—particularly in PTEN-deficient colorectal cancer (CRC). |
|
| DC70175 |
AEP07
Featured
|
AEP07 (AEP 07) is a selective small molecule inhibitor of PARP4 (vPARP) inhibitor with IC50 of 0.8 uM, >12-fold selective over other PARP family members. |
|
| DC70194 |
AMXI-5001 Hcl |
AMXI-5001 Hcl (AMXI 5001) is a novel, highly potent, orally active dual PARP1/2 (IC50 5/0.05 nM) and microtubule polymerization inhibitor, inhibits intracellular PAR formation with IC50 of 7 nM.AMXI-5001 binds to the catalytic domain of human PARP1 and is a weak tankyrase inhibitor (800-fold lower than IC50 towards either PARP1 or PARP2 enzymes).AMXI-5001 inhibited tubulin polymerization in a dose-dependent manner.AMXI-5001 exhibited selective antitumor cytotoxicity across a wide variety of human cancer cells with much lower IC50s than existing clinical PARP1/2 inhibitors.AMXI-5001 is highly active in both BRCA mutated and wild type cancers.AMXI-5001 elicited a remarkable In vivo preclinical anti-tumor activity in a BRCA mutated TNBC model induced complete regression of established tumors, including exceedingly large tumors, demonstrated superior anti-tumor effects compared to either single agent (PARP or microtubule) inhibitor or combination with both agents. |
|
| DC70198 |
And1 inhibitor CH3 |
And1 inhibitor CH3 is potent acidic nucleoplasmic DNA-binding protein 1 (And-1) inhibitor, reducess And‐1 expression level in IGROV1 cells with IC50 of 2.08 uM.CH3 induces acidic nucleoplasmic DNA‐binding protein 1 (And‐1) degradation via the E3 ubiquitin ligase CUL4B‐mediated proteasome degradation pathway.CH3 promotes the interaction between And‐1 and CUL4B by altering And‐1 conformation.CH3 exhibits the significant inhibition in a broad range of cancer cells in vitro and in vivo.CH3 (20 mg/kg and 40 mg/kg) reduced tumor growth of ovarian IGROV1 and breast MCF7 xenografts at both treated doses.CH3 also could overcome cisplatin resistance in ovarian cancer.And1 is an important factor for deoxyribonucleic acid (DNA) replication and repair, is overexpressed in many types of cancer but not in normal tissues. |
|