| DC70217 |
ASP9822 |
ASP9822 is a novel potent selective CDK7 inhibitor with IC50 of 4.3 nM, with anti-cancer activity. |

|
| DC70230 |
AZ5576
Featured
|
AZ5576 (AZ-5576) is a potent, selective CDK9 inhibitor with an IC 50 <5 nM, decreases phosphorlyation of Ser2-RNAPII in cells with an IC 50 of 96 nM.AZ5576 downregulated Mcl-1 and MYC protein abundance across multiple tested DLBCL cell lines and in primary DLBCL cells.MYC sensitizes DLBCL cells to AZ5576, genetic downregulation of MYC in U-2932 and OCI-LY19 cells resulted in diminished susceptibility to AZ5576.AZ5576 dose-dependently downregulated MYC transcription of a luciferase reporter containing multiple canonical MYC-MAX-binding sites, an effect more pronounced compared with multi-CDK inhibitor dinaciclib.AZ5576 restricts growth of xenografted DLBCL tumors in vivo. |

|
| DC70255 |
bio-THZ1 |
bio-THZ1 is a biotinylated version of THZ1 and binds irreversibly to CDK7. |
|
| DC70259 |
BLM inhibitor 2 |
BLM inhibitor 2 is a specific inhibitor of BLM (Bloom syndrome protein) ATPase-coupled DNA helicase activity (IC50=2.2 uM) via the allosteric binding site.BLM inhibitor 2 displays high selectivity over members of the RecQ-helicase family in ATP-turnover assays, including recombinant helicase domains (HD) from human WRN, human RecQ5 or E. coli helicase UvrD, and human RecQ1.BLM inhibitor 2 is a non-competitive inhibitor, does not interfere with ATP-binding, unliker ML216.BLM inhibitor 2 inhibits production of the single-stranded DNA product in a dose-dependent manner (IC50=1.8 uM) via inhibition of the helicase activity of BLM-HD.BLM (Bloom syndrome protein) is a RECQ-family helicase involved in the dissolution of complex DNA structures and repair intermediates (BLM, WRN, RECQ1, RECQ4, and RECQ5 in humans). |
|
| DC70283 |
CAM833 |
CAM833 (CAM-833) is a potent, specific chemical inhibitor of the RAD51-BRCA2 interaction and RAD51 oligomerization with Kd of 366 nM;
CAM833 inhibited RAD51 foci formation 6 h after exposure to 3 Gy IR, in a concentration-dependent manner with an IC50 of 6 uM, causeed a concentration-dependent decrease in RAD51 foci accompanied by increased DNA damage in A549 cells.
CAM833 inhibted RAD51 molecular clustering at DNA damage sites visualized by SMLM, suppressed homologous recombination and potentiated cell-cycle arrest.
CAM833 potentiated the growth suppressive effect of PARP1 inhibition in BRCA2 wild-type cells, as well as dose-dependent growth inhibition when combined with ionizing radiation. |
|
| DC70285 |
CaNDY |
CaNDY is a highly potent rectifier of the aberrant splicing, potently inhibits DYRK1A, DYRK1B, CLK1/CLK2/ with IC50 of 21.6/68.3/116 nM in in vitro kinase assays.CaNDY is a potent and selective inhibitor of DYRK family kinases, CaNDY induces selective degradation of DYRK1A, and inhibits catalytic activity of recombinant DYRK1A with IC50 value of 7.9 nM by competing with ATP, antagonizes the CDC37 interaction with DYRK1A.CaNDY demonstrated CFTR pseudo exon suppression with EC50 of 1.2 uM, 20.4 times lower than that of TG003.Splice modulation by CaNDY is mediated by its CLK-inhibition activity.CaNDY, but not CFTR modulator VX-809, rescues mature CFTR expression from the CFTR gene with the 3,849 + 10 kb C>T mutation, suppresses SRSF phosphorylation.CaNDY (3 uM) treatment rescued CFTR channel activity from CFTRC>T-GFP at an extent comparable to that of CFTRWT-GFP, demonstrating a restoration of functional CFTR expression. |
|
| DC70298 |
CDDD11-8
Featured
|
CDDD11-8 is a potent CDK9 inhibitor co-targeting FLT3-ITD with Ki values of 8 and 13 nM, respectively.CDDD11-8 displays excellent kinome selectivity in a panel of 369 human kinases.CDDD11-8 displays antiproliferative activity against leukemia cell lines, and particularly potent effects against MV4-11 and MOLM-13 cells, which are known to harbor the FLT3-ITD mutation and mixed lineage leukemia (MLL) fusion proteins.CDDD11-8 causes a robust tumor growth inhibition by oral administration in animal xenografts, induces tumor regression at dose of 125 mg/kg. |
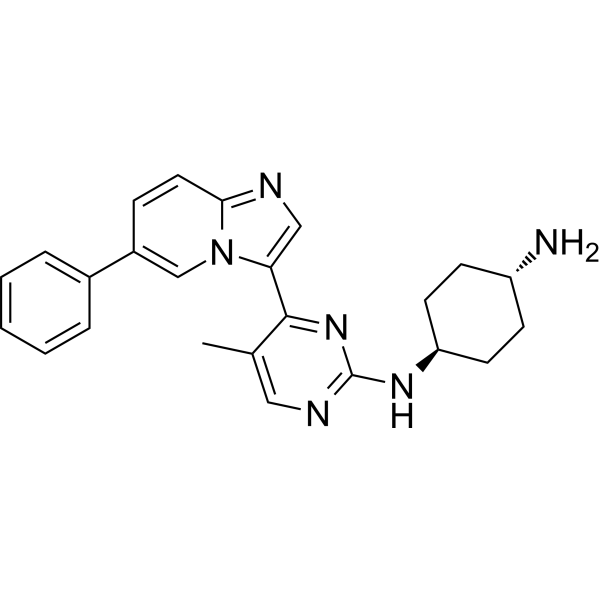
|
| DC70299 |
CDDD2-94 |
CDDD2-94 is a highly potent and selective CDK4 inhibitor with Ki of 2 nM, >140-fold selective for CDK4 over CDK6 (Ki=279 nM).CDDD2-94 is ineffective against other members of the CDK family, displays high selectivity against a panel of 369 human kinases at 1uM, with exceptionally selective-CLK, DYRKs and MYLK4 were the only kinases targeted potently.CDDD2-94 is the most selective CDK4 inhibitor identified to date.CDDD2-94 demonstrated antiproliferative activityagainst MV4-11 and MDA-MB-453 cell lines with GI50 of 0.107 and 0.325 uM respectively.CDDD2-94 inhibits S780-phosphorylated Rb (pRb(S780)) and decreases transcription of Rb1 and E2F-target genes in MDA-MB-453 cells.CDDD2-94 is well tolerated and efficacious in preclinical OC xenograft model, CDDD2-94 provides better safety profile than palbociclib towards the bone marrow. |
|
| DC70300 |
CFI-400945
Featured
|
CFI-400945 is a potent, highly selective PLK4 inhibitor with Ki of 0.26 nM and IC50 of 2.8 nM, does not significantly inhibits PLK1/2/3 at 50 uM; causes dysregulated centriole duplication, mitotic defects, and cell death in multiple cancer cell lines (A549 GI50=5 nM, OVCAR-3 GI50=18 nM); significantly inhibits human cancer xenografts; causes polyploidy, growth inhibition, and apoptotic death of murine and human lung cancer cells, despite expression of mutated KRAS or p53, produces supernumerary centrosomes and mitotic defects in lung cancer cells. |
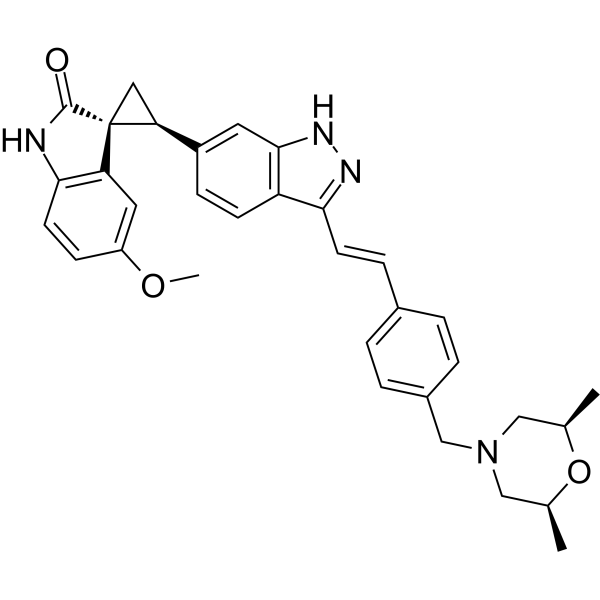
|
| DC70301 |
CFI-400945 fumarate |
CFI-400945 is a potent, highly selective, orally available PLK4 inhibitor with Ki of 0.26 nM and IC50 of 2.8 nM, does not significantly inhibits PLK1/2/3 at 50 uM; causes dysregulated centriole duplication, mitotic defects, and cell death in multiple cancer cell lines (A549 GI50=5 nM, OVCAR-3 GI50=18 nM); significantly inhibits human cancer xenografts; causes polyploidy, growth inhibition, and apoptotic death of murine and human lung cancer cells, despite expression of mutated KRAS or p53, produces supernumerary centrosomes and mitotic defects in lung cancer cells. |
|
| DC70309 |
cis-ISRIB
Featured
|
cis-ISRIB is a weak inhibitor of PERK signaling compared to trans-ISRIB , potently reverses the effects of eIF2α phosphorylation with IC50 of 600 nM.ISRIB reduces the viability of cells subjected to PERK-activation by chronic endoplasmic reticulum stress, displays significant enhancement in spatial and fear-associated learning.ISRIB is a potent inhibitor of the integrated stress response (ISR)integrated stress response (ISR). |
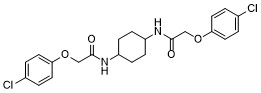
|
| DC70313 |
CLK4 inhibitor 96 |
CLK4 inhibitor 96 is a novel potent CLK4 inhibitor with IC50 of 57.5 nM. |
|
| DC70336 |
Cycloheximide |
Cycloheximide (Naramycin A, Actidione) is a widely-used protein synthesis inhibitor that inhibits protein biosynthesis in eukaryotic organisms with IC50 of 532.5 nM and 2880 nM for protein synthesis and RNA synthesis in vivo, respectively; block the elongation phase of eukaryotic translation, binds the ribosome and inhibits eEF2-mediated translocation; impairs memory more as footshock intensity increases, enhances memory in an inverted-U dose-response manner at low dose; also sensitizes COLO 205 cells to TNF-alpha-induced programmed cell death. |
|
| DC70350 |
Debio 0123
Featured
|
Debio 0123 (Debio0123) is a potent, orally available, and highly selective Wee1 kinase inhibitor with IC50 of 0.8 nM, Ki of 0.1 nM.Debio 0123 is highly selective to Wee1 on 465 selected kinases in a cellfree system, does not inhibit Plk1 and Plk2, as also reported in recent publications for AZD1775.Debio 0123 exhibits in vitro growth inhibition activity in a broad number of human cancer cell lines (IC50= 0.109 to 7.08 uM).Debio 0123 prevent CDC2 / Cdk1 phosphorylation, induces γH2AX and enhances phosphorylation of histone H3.Debio 0123 induces apoptosis through mitotic catastrophe following cell cycle progression despite accumulation of unrepaired DNA damage both in vitro and in vivo. |
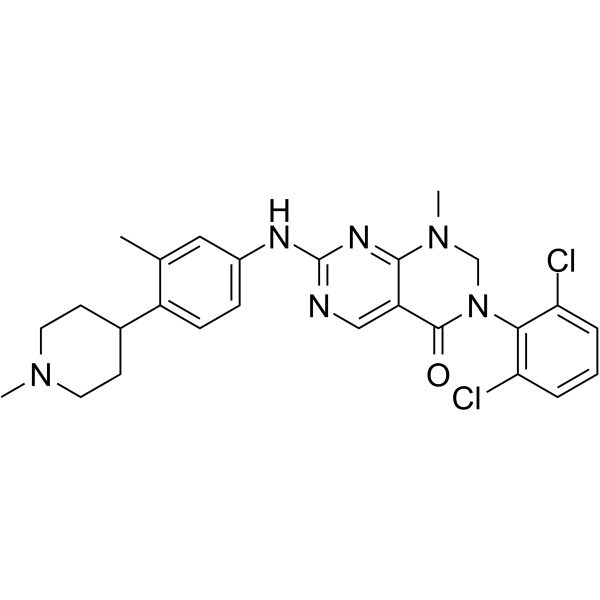
|
| DC70400 |
F114 |
F114 is a novel, first-in-class, dual aurora and lim kinase inhibitor with IC50 of 72 nM and 137 nM against Aurora-A and Limk1, respectively, F114 inhibits GBM proliferation and invasion. |
|
| DC70440 |
GPS167
Featured
|
GPS167 is a specific small molecule splicing regulator SRSF10 inhibitor, modulates BCLAF1 splicing with IC50 of 2 uM in human colorectal HCT116 cells, directly inhibits CLK1, CLK2 and CLK4, but not SRPK1 and DYRK1A.GPS167 promotes the dephoshorylation of SRSF10 and changes its interaction with partner proteins.GPS167 treatment leads to a partial dephosphorylation of SRSF10 and increases the recovery of CLK1 and CLK4.GPS167 impairs the growth of cancer cell line, elicits p53-dependent cytotoxicity.GPS167 is cytotoxic for human colorectal cancer organoids but not normal organoids. |
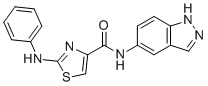
|
| DC70483 |
HH-N25 |
HH-N25 is a a potent and selective topoisomerase I (TOP1) inhibitor with potent antiproliferative activities against a panel of human breast cancer cell lines (IC50=0.045-4.2 uM).HH-N25 shows potent antitumor activities in human breast cancer cells in vitro and in vivo. |
|
| DC70514 |
IPP-14 |
IPP-14 is a novel PAK1 inhibitor that selectively blocks the PAK1-PUMA binding and suppresses cell proliferation via PUMA-dependent manner; suppresses cancer cell viability and migration, induces p21 expression and induces M-phase arrest by inhibition of PAK1; also induces p21/WAF1/CIP1 (Cyclin-dependent kinase inhibitor 1A) expression by releasing from Bcl-2 and by inhibition of AKT-mediated p21 suppression. |
|
| DC70557 |
Lapcin |
Lapcin is a potent dual topoisomerase I/II inhibitor with nM to pM IC50s against diverse cancer cell lines (Colon cancer HT29 cell IC50=0.516 nM);
Lapcin showed weak activity against DNA gyrase (IC50>20.5 uM).
While only weakly active against DNA gyrase, lapcin was a potent of inhibitor of both DNA relaxation by topoisomerase I (IC50=2.17 uM) and DNA decatenation by topoisomerase II (IC50=7.53uM), 14 times more potent than the alkaloid camptothecin.
Lapcin was more potent than the topoisomerase II inhibitor etoposide against all of the cell lines we tested, with the exception of the lung cancer cell NCI-H226, against which it was essentially equipotent.
Cell lines expressing wild-type p53 (HCT116, H226, MCF7) tended to show higher IC50s than p53 reduced cell lines (NCI-H1299, HT29, Colo205, Hela). |
|
| DC70567 |
LMP517 |
LMP517 (NSC 781517) is a non-camptothecin, dual TOP1 and TOP2 topoisomerase inhibitor.LMP517 showed better antitumor activity than its parent compound LMP744 against H82 (small cell lung cancer) xenografts. |
|
| DC70580 |
M3541
Featured
|
M3541 (M 3541) is a novel potent, selective and ATP-competitive ATM kinase inhibitor with IC50 of 0.25 nM (ATP, 10 uM).M3541 displays a good margin of selectivity (>60-fold) against the closely related PIKK family members with DNA-PK, PI3K isoforms, mTOR, and ATR.M3541 inhibits the phosphorylation of ATM as well as its cellular substrates CHK2, KAP1, and p53 in a concentration-dependent manner in A549 cells.M3541 inhibits double strand break repair and sensitizes cancer cells to radiation.M3541 suppresses ATM pathway activity and potentiates radiation efficacy in xenograft models of human cancer, synergizes with topoisomerase and PARP inhibitors. |
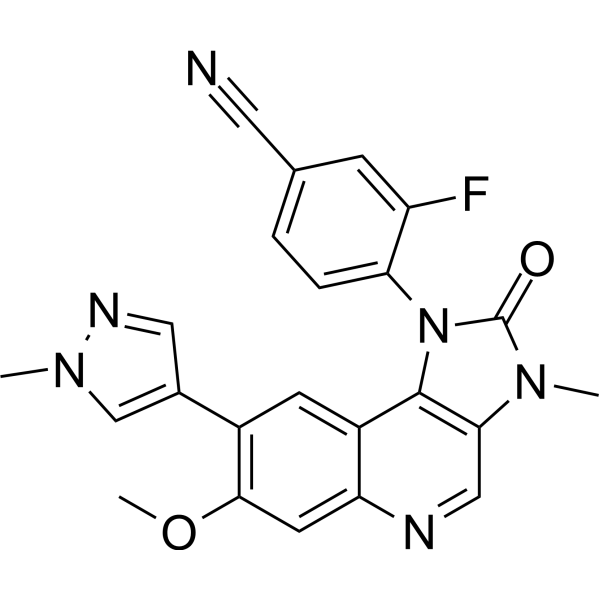
|
| DC70581 |
M4076 |
M4076 is a higly potent, selective, ATP-competitive inhibitor of ATM kinase with IC50 of 0.2 nM.M4076 sensitizes tumor cell lines to radiation therapy in vitro and strongly enhances the anti-tumor activity of ionizing radiation in vivo. |
|
| DC70595 |
MFH290
Featured
|
MFH290 (MFH-290) is a potent, highly selective, covalent inhibitor of CDK12/13 with IC50 of 25/49 nM.MFH290 forms a covalent bond with Cys-1039 of CDK12, and CDK12-dependent as mutation of Cys-1039 rendered the kinase refractory to MFH290.MFH290 exhibits excellent kinome selectivity, inhibits the phosphorylation of serine-2 in the C-terminal domain (CTD) of RNA-polymerase II (Pol II), and reduces the expression of key DNA damage repair genes.MFH290 restored Pol II CTD phosphorylation and DNA damage repair gene expression AND augments the antiproliferative effect of the PARP inhibitor olaparib. |
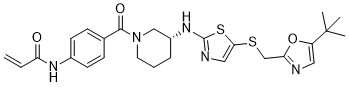
|
| DC70622 |
MSC 2711186 |
MSC2711186 is a potent, selective, cell active pan-SRPK with IC50 of 2.7/8.1/0.59 nM for SRPK1/2/3, repectively.MSC2711186 does not show any activity towards the CLKs, and is selective in an in vitro kinase panel from Reaction Biology at 1 µM against 395 Kinases.MSC2711186 displayed an IC50 of 98 nM on SRPK1 and 40 nM on SRPK3 in intact cells and 44 nM on SRPK1, 149 nM on SRPK2 and 40nM on SRPK3 in lysed cells in NanoBRETTM assay.Serine-arginine protein kinases (SPRKs) are a subfamily of serine-threonine kinases, regulating pre-mRNA splicing in response to extracellular stimuli by phosphorylating serine/arginine (SR)-rich splicing factors. The human genome encodes three SRPK genes, SRPK1, SRPK2 and SRPK3. While SRPK1 has been detected in many human tissues at varying expression levels, SRPK2 and SRPK3 exhibit a more tissue-specific expression. |
|
| DC70667 |
NU5455 |
NU5455 (NU-5455) is a potent, highly selective, oral inhibitor of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) activity with IC50 of 8.2 nM.NU5455 displays selectivity versus Vps34 (8.7-fold), PI3Kδ (33.7-fold), ATM/ATR (both >1200-fold), 228-fold selectivity margin for DNA-PKcs kinase activity versus that of PI3Kα.NU5455 inhibited radiation-induced activation of DNA-PK with an IC50 of 168 nM, but did not inhibit IGF-stimulated activation of AKT in MCF7 cells even at 10 uM.NU5455 inhibited the repair of DNA-DSBs induced by treatment with enzyme AfeI or ScaI restriction endonucleases within a 24-hour period in HEK293T cells, significantly increased the number of colocalized γH2AX and 53BP1 foci.NU5455 is a highly selective inhibitor of DNA-PKcs that is active in cells and that can perturb DNA-DSB repair by NHEJ.NU5455 preferentially augmented radiotherapy in subcutaneous Calu-6 and A549 lung tumor xenografts, increased both the efficacy and the toxicity of a parenterally administered topoisomerase inhibitor, NU5455 enhanced the activity of doxorubicin released locally in liver tumor xenografts without inducing any adverse effect. |
|
| DC70678 |
PARPYnD |
PARPYnD is the first cell-active photoaffinity probe (AfBP) for PARP protein class, based on triple PARP1/2/6 inhibitor AZ9482.PARPYnD is a robust tool for profiling PARP1/2 and is used to profile clinical PARP inhibitor olaparib, identifying several novel off-target proteins.PARPYnD can enrich recombinant PARP6 spiked into cellular lysates and inhibits PARP6 in cell-free assays, it does not label PARP6 in intact cells.PARPYnD inhibits PARP1/2/6 with IC50 of 38/6/230 nM, respectively. |
|
| DC70687 |
PF-06842874 |
PF-06842874 is a potent, selective CDK4/6 inhibitor. |
|
| DC70743 |
RP12146 |
RP12146 (RP12146) is a novel, selective, and potent small molecule inhibitor of PARP1/2 with IC50 of 0.6/0.5 nM, with several fold selectivity over other isoforms.RP12146 inhibited cell growth in both BRCA mutant and non-BRCA mutant cancer cell lines with a GI50 range of 0.043 to 19.83 µM in a dose dependent manner.RP12146 demonstrated anti-tumor activities in both tumor size and tumor weight in OVCAR3 xenograft model. |
|
| DC70763 |
SCR7 |
SCR7 is a specific inhibitor of nonhomologous end-joining (NHEJ), blocks Ligase IV-mediated joining by interfering with its DNA binding but not that of T4 DNA Ligase or Ligase I; inhibits NHEJ in a Ligase IV-dependent manner within cells, causes accumulation of double-strand breaks (DSBs), and activates the intrinsic apoptotic pathway; decreases cell proliferation of MCF7, A549, and HeLa with IC50 of 40, 34, and 44 uM, respectively; increases the efficacy of DSB-inducing therapeutic modalities in mouse xenografts; increases the efficiency of precise genome editing with CRISPR-Cas9 in mammalian cell lines and in mice. |
|
| DC70766 |
Senexin C
Featured
|
Senexin C is a novel potent, selective and orally bioavailable CDK8/19 inhibitor with Kd of 1.4 and 2.9 nM for CDK8/CycC and CDK19/CycC, respectively.Senexin C inhibits CDK8/CycC with IC50 of 3.6 nM, shows high selectivity against other HDAC isoforms.Senexin C is more metabolically stable and provides a more sustained inhibition of CDK8/19-dependent cellular gene expression when compared with the prototype inhibitor Senexin B.Senexin C inhibits MV4-11 leukemia growth in a systemic in vivo model with good tolerability. |
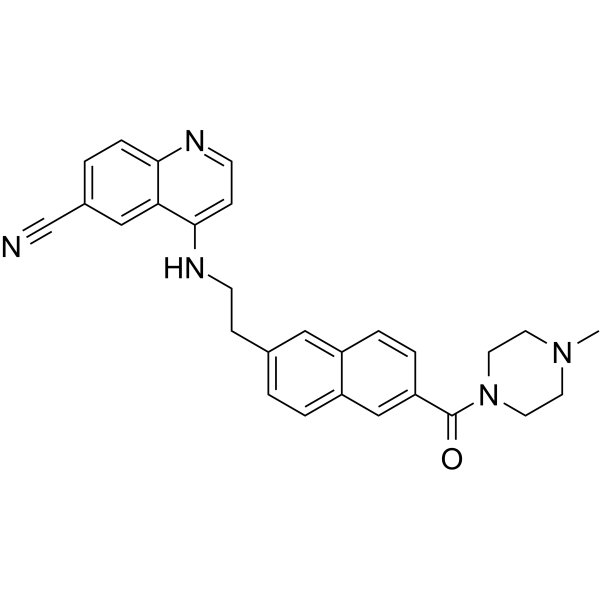
|