| DC70819 |
SY-5102 |
SY-5102 (SY5102) is a highly potent, selective, noncovalent, orally available CDK7 inhibitor with SPR Kd of 0.03 nM (binding to CDK7/Cyclin H).SY-5102 (SY5102) displays highly selectivity against CDK2, CDK9, and CDK12 (all IC50>75 nM).SY-5102 potently inhibits HCC70 cell proliferation with EC50 of 9 nM.SY-5102 showed dose-dependent tumor growth inhibition including tumor regression at 4 mg/kg po (dosed twice daily) in an HCC70 cell line derived xenograft mouse model. |

|
| DC70839 |
TH9028 |
TH9028 (TH-9028) is a potent and selective inhibitor of folate metabolism enzyme MTHFD2 with IC50 of 11 nM.TH9028demonstrated efficacy on HL-60 cell viability with EC50 of 11 nM.TH9028 potently inhibits related MTHFD proteins MTHFD2L and MTHFD1-(DC) with IC50 of 47 and 16 nM, respectively. |
|
| DC70840 |
TH9619
Featured
|
TH9619 (TH-9619) is a potent and selective inhibitor of folate metabolism enzyme MTHFD2 with IC50 of 47 nM.TH9619 demonstrated efficacy on HL-60 cell viability with EC50 of 11 nM.TH9619 potently inhibits related MTHFD proteins MTHFD2L and MTHFD1-(DC) with IC50 of 47 and 16 nM, respectively.TH9619 displays high potency and cancer selectivity in AML models. induce thymine-less DNA damage and apoptosis, causes uracil misincorporation into DNA, sensitize cancer cells to ATR-signaling blockade.TH9619 impairs cancer progression in vivo.TH9619 displays high selectivity toward binding and stabilizing MTHFD2 over other common folate metabolism targets such as DHFR, thymidylate synthase (TYMS), serine hydroxymethyltransferase (SHMT) 1 and SHMT2. |
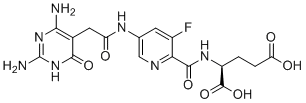
|
| DC70844 |
Thymidine Phosphorylase inhibitor 8g |
Thymidine Phosphorylase inhibitor 8g (hTP inhibitor 8g) is a novel potent, selective inhibitor of human thymidine phosphorylase (hTP) with IC50 of 0.12 uM; induces DNA damage measured with the alkaline comet assay in HepG2 cells; hTP inhibitor 8g (50 mg/kg/day) for two weeks (5 days/week) significantly reduced tumor growth using an in vivo glioblastoma model. |
|
| DC70903 |
Wee1 Inhibitor II |
Wee1 Inhibitor II is a potent, selective, ATP-binding site-targeting inhibitor of Wee1 with IC50 of 59 nM, 590-fold selectivity over the related checkpoint kinase Chk1 (IC50=35 uM). |
|
| DC70930 |
Zebularine |
Zebularine (NSC309132, 4-Deoxyuridine) is a nucleoside analog of cytidine and inhibitor of cytidine deaminase, also inhibits DNA methylation and tumor growth both in vitro and in vivo. |
|
| DC70958 |
4-Aminonaphthalimide |
4-Aminonaphthalimide is a potent PARP inhibitor and potentiates the cytotoxicity of γ-radiation in cancer cells. |
|
| DC70984 |
(S)-CR8
Featured
|
(S)-CR8 is the S-isomer of CR8. (S)-CR8 is a potent and selective CDK inhibitor with IC50s of 0.060, 0.080, 0.11, 0.12, and 0.15 μM for CDK2/cyclin E, CDK2/cyclin A, CDK9/cyclin T, CDK5/p25, and CDK1/cyclin B, respectively. (S)-CR8 reduces SH-SY5Y cells survival (IC50 0.40 μM). |
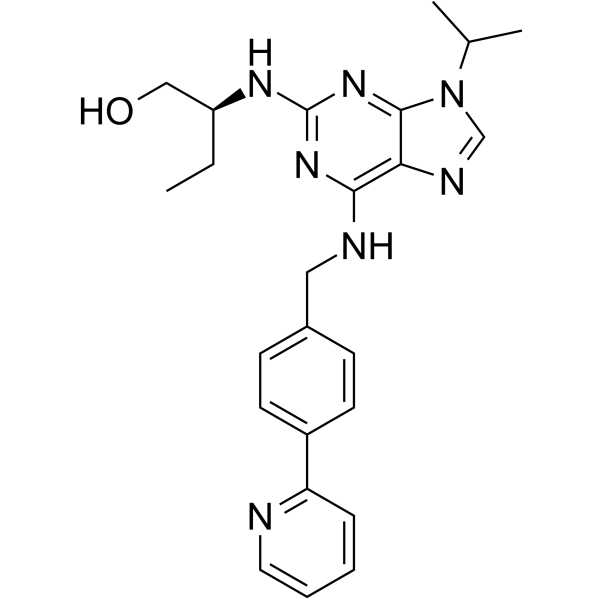
|
| DC71000 |
Alatrofloxacin |
Alatrofloxacin, the parenteral prodrug of Trovafloxacin, is a fluoronaphthyridone which contains an L-alanyl-L-alanyl salt. Alatrofloxacin functions similar to other fluoroquinolone antibiotics in that it not only has antibiotic activity to kill invading organisms by interfering with DNA synthesis, it possesses immunosuppressive activity. |
|
| DC71005 |
AS-136A |
AS-136A is an orally active non-nucleoside inhibitor of the measles virus RNA-dependent RNA polymerase (RdRp) with an IC50 of 2 µM for measles virus. |
|
| DC71122 |
TDRL-X80 |
TDRL-X80 is a potent inhibitor of xeroderma pigmentosum group A (XPA) protein. TDRL-X80 inhibits XPA’s DNA binding activity. TDRL-X80 exhibits activity against single, double, and Cisplatin-damaged DNA with IC50s of 18, 20, and 29 μM in fluorescence polarization (FP) analyses , and with IC50s of 21, 39, and 28 in ELISA Analysis. |
|
| DC71211 |
AZD5099 |
AZD5099, an antibacterial agent, is a potent and selective bacterial topoisomerase II inhibitor. AZD5099 potently inhibits the infections caused by Gram-positive and fastidious Gram-negative bacteria. |
|
| DC71215 |
ZDLD20 |
ZDLD20, a β-carboline, is orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 6.51 μM. ZDLD20 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cell cycle. ZDLD20 exhibits potent anticancer activity. |
|
| DC71226 |
Lurtotecan |
Lurtotecan (GI147211; OSI-211), a semisynthetic Camptothecin analog, is a topoisomerase I inhibitor. Lurtotecan has anticancer effects. |
|
| DC71231 |
ZDLD13 |
ZDLD13, a β-carboline, is an orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 0.38 μM. ZDLD13 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cell cycle. ZDLD13 shows significant tumor growth inhibition in HCT116 tumor xenograft model. |
|
| DC71239 |
6-Mercaptopurine-13C2,15N |
6-Mercaptopurine-13C2,15N (Mercaptopurine-13C2,15N) is the 13C- and 15N-labeled 6-Mercaptopurine. 6-Mercaptopurine is a purine analogue which acts as an antagonist of the endogenous purines and has been widely used as antileukemic agent and immunosuppressive drug. |
|
| DC71298 |
Zn(BQTC) |
Zn(BQTC) is a highly potent mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) inhibitor. Zn(BQTC) causes severe damage to the mtDNA and nDNA, sequentially disruptes mitochondrial and nuclear functions. Zn(BQTC) promotes the DNA damage-induced apoptotic signaling pathway. Zn(BQTC) has selectively antiproliferative activity against A549R cells. Zn(BQTC) can be used for researching anticancer. |
|
| DC71299 |
Nogalamycin |
Nogalamycin is an anthracyclinone antibiotic. Nogalamycin is a potent antibiotic against Gram-positive bacteria, also has cytotoxicity against certain tumor cells. Nogalamycin is produced by Streptomyces nogalater var. Nogalater. Nogalamycin selectively inhibits RNA synthesis after binding to DNA template. Nogalamycin can be used for researching anticancer. |
|
| DC71300 |
Branaplam hydrochloride
Featured
|
Branaplam (LMI070; NVS-SM1) hydrochloride is a highly potent, selective and orally active survival motor neuron-2 (SMN2) splicing modulator with an EC50 of 20 nM for SMN. Branaplam hydrochloride inhibits human-ether-a-go-go-related gene (hERG) with an IC50 of 6.3 μM. Branaplam hydrochloride elevates full-length SMN protein and extends survival in a severe spinal muscular atrophy (SMA) mouse model. |

|
| DC71301 |
Pencitabine |
Pencitabine (Pen) is an orally active anticancer agent. Pencitabine interferes with DNA synthesis and function by inhibiting multiple nucleotide-metabolizing enzymes and by misincorporation into DNA. |
|
| DC71302 |
UIAA-II-232 |
UIAA-II-232 (compound 19b) is a potent DNA gyrase catalytic inhibitor with an IC50 value of 3.5 µM. |
|
| DC71303 |
DPQ |
DPQ is a potent PARP-1 inhibitor. DPQ can reduce the N-methyl-d-aspartate (NMDA)-induced PARP activation, restoring ATP to near control levels and significantly attenuating neuronal injury in the severe NMDA exposure model. DPQ can be used for researching neuroprotection. |
|
| DC71304 |
SK-575-NEG |
SK-575-NEG (compound 28), a methylation counterpart of SK-575, is synthesized by methylation of the amino group of piperidine-2,6-dione in SK-575 as an control compound. SK-575-NEG is strongly bound to PARP1, with an IC50 of 2.64 nM. SK-575-NEG was completely ineffective in inducing PARP1 degradation in MDA-MB-436 and Capan-1 cells at concentrations up to 1 μM. |
|
| DC71305 |
SDOX |
SDOX is the Doxorubicin (DOX) prodrug. The loaded DOX prodrugs (SDOX) which can release the parent drugs DOX triggered by excessive GSH in tumor cells, minimize the unexpected side effects on normal tissues without compromising the potency. |
|
| DC71306 |
Berberine sulfate |
Berberine sulfate is an alkaloid isolated from the Chinese herbal medicine Huanglian, as an antibiotic. Berberine sulfate induces reactive oxygen species (ROS) generation and inhibits DNA topoisomerase. Berberine sulfate has antineoplastic properties. |
|
| DC71307 |
LEB-03-146 |
LEB-03-146 is a WEE1 DUBTAC (deubiquitinase-targeting chimera) linking AZD1775 (Adavosertib) to the OTUB1 recruiter EN523 through a PEG2 linker. LEB-03-146 shows significant WEE1 stabilization in HEP3B hepatoma cancer cells. |
|
| DC71491 |
Methotrexate monohydrate |
Methotrexate (Amethopterin) monohydrate, an antimetabolite and antifolate agent, inhibits the enzyme dihydrofolate reductase, thereby preventing the conversion of folic acid into tetrahydrofolate, and inhibiting DNA synthesis. Methotrexate monohydrate, also an immunosuppressant and antineoplastic agent, is used for the research of rheumatoid arthritis and a number of different cancers (such as acute lymphoblastic leukemia). |
|
| DC71492 |
ddCTP trisodium |
ddCTP trisodium is one of 2',3'-dideoxyribonucleoside 5'-triphosphates (ddNTPs) that acts as chain-elongating inhibitor of DNA polymerase for DNA sequencing. ddCTP trisodium is a nucleoside analog that targets the reverse transcriptase of human immunodeficiency virus (HIV). ddCTP trisodium can be used for AIDS research. |
|
| DC71493 |
Basroparib |
Basroparib is a potent poly (ADP-ribose) polymerase (PARP) inhibitor, with antineoplastic activity. |
|
| DC71494 |
TIQ-A |
TIQ-A is a potent TNKS (poly-ART, PARP) inhibitor, with an IC50 of 24 nM for TNKS2. TIQ-A is a potential anti-ischemic agent. |
|