 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC70279 | BY27 |
BY27 (BD2 inhibitor BY27) is a potent, highly selective BET BD2 inhibitor with Ki of 8.9/8.2/14.2 nM for BRD2/3/4-BD2 respectively.BY27 displays 38-fold selectivity for BRD2-BD2 over BRD2-BD1.BY27 showed good antitumor activity in the MV4-11 xenograft model with an improved in vivo tolerance in mice compared with I-BET762.
More description
|

|
| DC70271 | BRD8 inhibitor DN02 |
BRD8 inhibitor DN02 is a first-in-class selective and cellularly active probe for NuA4 factor BRD8 (BD1) with IC50 of 34 nM, no affinity for BRD8 {BD2}, CBP, and p300.
More description
|
|
| DC70270 | BRD4 D1 inhibitor 30 |
BRD4 D1 inhibitor 30 is a high-affinity, BRD4 D1-selective chemical probe with Kd of 18 nM, 500-fold selectivity against BRD2 D1 and BRD4 D2.BRD4 D1 inhibitor 30 down regulated inflammation, IL-8 and chemokine in cell based assays, but was unable to reduce c-Myc expression at low concentration in multiple myeloma cells.
More description
|
|
| DC70269 | BR-001 |
BR-001 (BR001) is a potent, selective, allosteric inhibitor of EED subunit of PRC2 complex, disrupts EED-H3K27me3 interaction (IC50=4.5 nM).BR-001 directly binds to EED in the H3K27me3-binding pocket, BR-001 significantly increases the thermal stability of EED in thermal shift assay.BR-001 is selective for EED, has no activity against 371 wild-type kinases.BR-001 significantly reduced the cellular H3K27me3 level and also exhibited a strong antiproliferative effect in Karpas 422 cells.BR-001 inhibited proliferation of DLBCL cells and tumor growth, and upregulated target genes expression.BR-001 has potent antitumor efficacy in vivo, modulates immune response to suppress syngeneic CT26 colon tumor.
More description
|
|
| DC70238 | BAY-155 |
BAY-155 (BAY155) is a novel, potent and selective menin-MLL inhibitor with binding IC50 of 8 nM, 10-fold better compared to that of MI-503;
BAY-155 demonstrated a significantly enhanced selectivity profile compared to MI-503 in a panel of assays covering numerous safety pharmacology-relevant targets including GPCRs, ion channels and transporters.
BAY-155 shows anti-proliferative activity in a large cell line panel, exhibits specific therapeutic activityin AML/ALL models.
More description
|
|
| DC70184 | ALKBH5 inhibitor 6 |
ALKBH5 inhibitor 6 is a small molecule inhibitor of RNA 6-N-methyladenosine (m6A) demethylase ALKBH5, inhibits ALKBH5 RNA m6A demethylation activity with IC50 of 1.79 uM.
More description
|
|
| DC70183 | ALKBH5 inhibitor 3 |
ALKBH5 inhibitor 3 is a small molecule inhibitor of RNA 6-N-methyladenosine (m6A) demethylase ALKBH5, inhibits ALKBH5 RNA m6A demethylation activity with IC50 of 0.84 uM.
More description
|
|
| DC70178 | AH237 |
AH237 (AH-237) is a potent and selective inhibitor for PRMT4 and PRMT5 with IC50 of 2.8 and 0.42 nM, respectively.AH237 (AH-237) displayed over 50-fold selectivity against a panel of 41 MTases such as PRMTs, PKMTs, NTMT1/2, and DNA methyltransferases (DNMTs), Its potency was also confirmed for PRMT1 (IC50 = 5.9 uM) and PRMT7 (IC50 = 831 nM).
More description
|
|
| DC70136 | TAF1 bromodomain inhibitor |
A potent and selective TAF1(2) bromodomain inhibitor.
More description
|
|
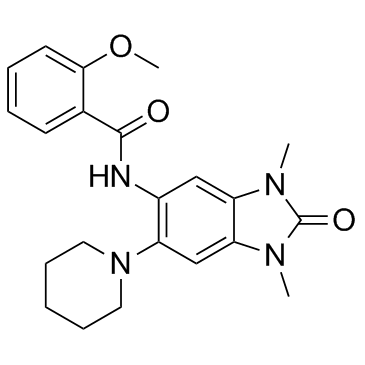
| DC8360 | GSK 5959 Featured |
GSK 5959 is a potent, cell-permeable inhibitor of the BRPF1 bromodomain (IC50 = 80 nM).
More description
|
|
| DC50236 | MC4355 |
MC4355 is a dual inhibitor of EZH2 and histone deacetylase (HDAC).
More description
|
|
| DC50234 | (R)-HH2853 |
(R)-HH2853 is a mutant EZH2 inhibitor with an IC50 of <100 nM for EZH2-Y641F. (R)-HH2853 can be used for cancer and autoimmune diseases (WO2018045971A1; compound 201).
More description
|
|
| DC50233 | (S)-HH2853 |
(S)-HH2853 (compound 200), a PYRIDINO five membered aromatic ring compound, is a potent EZH1/2 dual inhibitor with an IC50 of <100 nM for EZH2_Y641F. (S)-HH2853 has the potential to be used in the research of anti-tumor or autoimmune diseases.
More description
|
|
| DC50211 | TFMB-(S)-2-HG |
TFMB-(S)-2-HG is a potent inhibitor of the 5'-methylcytosine hydroxylase TET2. TFMB-(S)-2-HG also inhibits the EglN prolyl hydroxylases. TFMB-(S)-2-HG has the potential for the research of acute myeloid leukemia (AML).
More description
|
|
| DC49605 | CREB-IN-1 TFA |
CREB-IN-1 TFA is a potent, orally active CREB inhibitor (IC50=0.18 µM). CREB-IN-1 TFA inhibits breast cancer cell growth.
More description
|
|
| DC49603 | BRD4 Inhibitor-16 |
BRD4 Inhibitor-16 (Compound 4) is a potent inhibitor of bromodomain 4 (BRD4). Overexpression of bromodomain 4 (BRD4) is closely correlated with a variety of human cancers by regulating the histone post-translational modifications. BRD4 Inhibitor-16 represents a useful tool for explorative studies of BRD4 inhibition, such as an improved understanding of BRD4 inhibitor release-related information.
More description
|
|
| DC49602 | BRD4 Inhibitor-17 |
BRD4 Inhibitor-17 (Compound 5i) is a potent inhibitor of BRD4 with an IC50 of 0.33 μM. BRD4 Inhibitor-17 plays crucial role in regulating transcription of inflammatory, proliferation and cell cycle genes. BRD4 Inhibitor-17 serves as potential antidotes for arsenicals.
More description
|
|
| DC49600 | PARP1/BRD4-IN-1 |
PARP1/BRD4-IN-1 is a potent and high selective PARP1/BRD4 inhibitor (IC50s of 49 and 202 nM in PARP1 and BRD4, respectively). PARP1/BRD4-IN-1 represses the expression and activity of PARP1 and BRD4 to synergistically inhibit the malignant growth of pancreatic cancer cells.
More description
|
|
| DC49599 | (R,S)-CFT8634 |
(R,S)-CFT8634 is a selective and orally active BRD9 protein degrader. (R,S)-CFT8634 has the potential for the research of disorders mediated by BRD9, including but not limited to abnormal cellular proliferation (extracted from patent WO2021178920A1, compound 176).
More description
|
|
| DC49598 | (R,R)-CFT8634 |
(R,R)-CFT8634 is a selective and orally active BRD9 protein degrader. (R,R)-CFT8634 has the potential for the research of disorders mediated by BRD9, including but not limited to abnormal cellular proliferation (extracted from patent WO2021178920A1, compound 175).
More description
|
|
| DC49597 | FHT-1204 |
FHT-1204 is a potent SMARCA4/SMARCA2 ATPase (BRG1 and BRM) inhibitor with IC50s of ≤10 nM (WO2020160180A1; compound 70).
More description
|
|
| DC49596 | FHT-1015 |
FHT-1205 is a potent SMARCA4/SMARCA2 ATPase (BRG1 and BRM) inhibitor with IC50s of ≤10 nM (WO2020160180A1; compound 67).
More description
|
|
| DC49595 | BRM/BRG1 ATP Inhibitor-2 |
BRM/BRG1 ATP Inhibitor-2 is a BRG1/BRM ATPase inhibitor for the treatment of BAF-related disorders.
More description
|
|
| DC49594 | Sirt2-IN-5 |
Sirt2-IN-5 is a potent SIRT2 inhibtor.
More description
|
|
| DC49593 | AGK7 |
AGK7 is a potent inhibitor of sirtuin 2 (SIRT2). AGK7 rescues alpha-synuclein toxicity and modified inclusion morphology in a cellular model of Parkinson's disease. AGK7 protects against dopaminergic cell death both in vitro and in a Drosophila model of Parkinson's disease.
More description
|
|
| DC49592 | LSD1-IN-14 |
LSD1-IN-14 is a potent and selective LSD1 inhibitor (IC50=0.89 μM). LSD1-IN-14 can significantly inhibit the proliferation of A549 and THP-1 cells and induce the apoptosis of tumor cells.
More description
|
|
| DC49591 | NSC636819 |
NSC636819 is a competitive and selective inhibitor of KDM4A/KDM4B. KDM4A/KDM4B are potential progression factors for prostate cancer. NSC636819 has the potential for the research of cancer diseases, especially prostate cancer.
More description
|
|
| DC49590 | MC4343 |
MC4343 is a potent and dual inhibitor of EZH2 and histone deacetylase. MC4343 has the potential for the research of cancer disease.
More description
|
|
| DC49589 | KD 5170 |
KD 5170 is a pan inhibitor of histone deacetylases (HDACs) and exhibits broad spectrum antitumor activity in vitro and in vivo.
More description
|
|
| DC49588 | HDAC-IN-30 |
HDAC-IN-30 is a novel multi-target HDAC inhibitor, including HDAC1 (IC50=13.4 nM),HDAC2 (IC50=28.0 nM), HDAC3 (IC50=9.18 nM), HDAC6 (IC50=42.7 nM), HDAC8 (IC50=131 nM). HDAC-IN-30 exhibits potent antitumor efficacy.
More description
|
|