 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC49634 | GLP-1R agonist 8 |
GLP-1R agonist 8 is a potent GLP-1R agonist with an EC50 of < 2 nM (WO2021219019A1, compound 129a).
More description
|

|
| DC49633 | BMS-193884 |
BMS-193884 is a selective, orally active, and competitive ETA antagonist with 10000-fold greater affinity for the human ETA receptor (Ki=1.4 nM) than for the ETB receptor.
More description
|
|
| DC49632 | Dopamine D3 receptor ligand-1 |
Dopamine D3 receptor ligand is a potent, selective and high affinity ligand for Dopamine D3 receptor with 89-fold selective for D3 over D2 (D3 Ki= 8 nM, D2 Ki= 715 nM).
More description
|
|
| DC49631 | Ropinirole |
Ropinirole (SKF 101468) is an orally active, potent D3/D2 receptor agonist with a Ki of 29 nM for D2 receptor. Ropinirole has pEC50s of 7.4, 8.4 and 6.8 for hD2, hD3 and hD4 receptors, respectively. Ropinirole has no affinity for the D1 receptors. Ropinirole has the potential for Parkinson's disease.
More description
|
|
| DC49630 | Nemonapride |
Nemonapride is a highly potent dopamine D2 receptor antagonist with a Ki of 0.06 nM. Nemonapride also activates 5-HT1A receptor with an IC50 of 34 nM. Nemonapride is an antipsychotic that readily passes through the blood brain barrier and exhibits potent neuroleptic effects in animals.
More description
|
|
| DC49628 | CB65 |
CB65 is a potent and high affinity CB2 selective agonist with a Ki value of 3.3 nM. CB65 exhibits a Ki of >1000 nM for CB1 receptor.
More description
|
|
| DC49626 | Palmitoyl serinol |
Palmitoyl serinol (N-Palmitoyl serinol) is an analog of the endocannabinoid N-palmitoyl ethanolamine (PEA). Palmitoyl serinol improves the epidermal permeability barrier in both normal and inflamed skin.
More description
|
|
| DC49625 | Metazosin |
Metazosin (Kenosin) is a potent α1 adrenoceptor blocker. Metazosin is an antihypertensive agent lowering blood pressure.
More description
|
|
| DC49624 | A55453 |
A55453 is a prazosin analogue and a potent α1-adrenergic antagonist. 125I-A55453 is a high-affinity alpha 1-adrenergic receptor probe.
More description
|
|
| DC49623 | Bevantolol |
Bevantolol is a selective β-1 adrenoceptor antagonist. Bevantolol can be used for the research of angina pectoris and hypertension.
More description
|
|
| DC49621 | Prazobind |
Prazobind (SZL 49), a prazosin analog, is a potent alpha 1-adrenoceptor blocker. Prazobind competes for alpha 1-adrenoceptor binding sites with a similar potency (IC50=1 nM) in tissues enriched in both the alpha 1A (hippocampus) and alpha 1B (liver) subtypes.
More description
|
|
| DC49620 | A1AR antagonist 1 |
A1AR antagonist 1 (compound 18g) is a potent A1 adenosine receptor (AR) antagonist with Kis of 2.08, 6.91, and 31.2 nM for hA1, hA2A and hA2B, respectively.
More description
|
|
| DC49619 | PSB36 |
PSB36 is a potent and selective antagonist of adenosine A1 receptor, with Kis 0.12 nM, 187 nM, 552 nM, 2300 nM, and 6500 nM for rA1, hA2B, rA2A, hA3 and rA3 receptors respectively. PSB36 can be used for the research of hyperalgesia.
More description
|
|
| DC49618 | VUF-5574 |
VUF-5574 is a selective antagonist of adenosine A3 receptor with a Ki of 4.03 nM for the recombinant human receptor.
More description
|
|
| DC49617 | Adenosine receptor antagonist 2 |
Adenosine receptor antagonist 2 is an orally active A2a/A2b adenosine receptor antagonist with IC50s of 1 nM and 3 nM, respectively. Adenosine receptor antagonist 2 has anti-tumor activity.
More description
|
|
| DC49616 | A2A/A1 AR antagonist-1 |
A2A/A1 AR antagonist-1 (compound 1a) is dual potent A2A/A1 AR antagonist with Kis of 5.58 and 24.2 nM, respectively. A2A/A1 AR antagonist-1 has the potential for the research of ischemic stroke.
More description
|
|
| DC49615 | IHCH-3064 |
IHCH-3064 is a dual-acting compounds targeting Adenosine A2A Receptor and HDAC. IHCH-3064 exhibits potent binding to A2AR (Ki=2.2 nM) and selective inhibition of HDAC1 (IC50=80.2 nM), with good antiproliferative activity against tumor cell lines in vitro. IHCH-3064 is a tumor immunotherapeutic agent.
More description
|
|
| DC49614 | Adenosine receptor antagonist 3 |
Adenosine receptor antagonist 3 is a potent antagonist of adenosine receptor. Adenosine receptor antagonist 3 has the potential for the research of cancer disease (extracted from patent WO2019233994A1, compound 1).
More description
|
|
| DC49613 | MRS1334 |
MRS1334 is a potent and selective human adenosine A3 receptor antagonist with Kis of 2.69 nM, >100 nM, >100 nM for hA3, rA1, rA2A, respectively. MRS1334 blocks the protective effect of Cl-IB-MECA leading to significant bradycardia and elevated ST segment.
More description
|
|
| DC49612 | A1AR antagonist 2 |
A1AR antagonist 2 (compound 18h) is a potent A1 adenosine receptor (AR) antagonist with Kis of 1.49, 10.2, and 50.1 nM for hA1, hA2A and hA2B, respectively.
More description
|
|
| DC49611 | 5-HT6/5-HT2AR antagonist-1 |
5-HT6/5-HT2AR antagonist-1 is a potent dual 5-HT6/5-HT2AR antagonist with Ki values of 11 nM and 39 nM, respectively.
More description
|
|
| DC49610 | Gentisein |
Gentisein (NSC 329491), the major metabolite of Mangiferin, shows the most potent serotonin uptake inhibition with an IC50 value of 4.7 µM.
More description
|
|
| DC49609 | MS 245 oxalate |
MS 245 oxalate is a potent antagonist of 5-HT6 receptor with a Ki of 2 nM.
More description
|
|
| DC49607 | CGS 12066 dimaleate |
CGS 12066 (dimaleate) dimaleate is a selective 5-HT1B receptor agonist with an IC50 of 51 nM.
More description
|
|
| DC49606 | Sumatriptan |
Sumatriptan (GR 43175 free base) is an orally active 5-HT1 receptor agonist with Kis of 17 nM, 27 nM and 100 nM for 5-HT1D, 5-HT1B and 5-HT1A receptors, respectively. Sumatriptan can be used for migraine headache research.
More description
|
|
| DC49415 | CKLF1-C27 TFA |
CKLF1-C27, a C-terminal peptide of CKLF1, binds to CCR4 receptor and activates ERK1/2 pathway. CKLF1-C27 can abrogate the effect of CKLF1 on cells by competing for CCR4 receptor. CKLF1-C27 shows great effect on promoting proliferation on HUVECs. CKLF1-C27 has the potential for psoriasis research.
More description
|
|
| DC49414 | CKLF1-C27 |
CKLF1-C27, a C-terminal peptide of CKLF1, binds to CCR4 receptor and activates ERK1/2 pathway. CKLF1-C27 can abrogate the effect of CKLF1 on cells by competing for CCR4 receptor. CKLF1-C27 shows great effect on promoting proliferation on HUVECs. CKLF1-C27 has the potential for psoriasis research.
More description
|
|
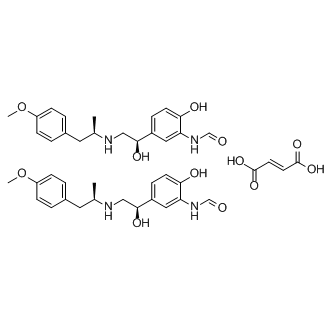
| DC9333 | Formoterol (Fumarate) Featured |
Formoterol fumarate(Foradil) is a potent, selective and long-acting β2-adrenoceptor agonist.
More description
|
|
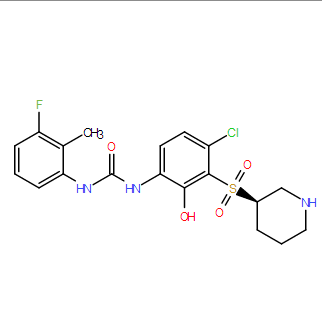
| DC9689 | Danirixin (GSK1325756) Featured |
Danirixin(GSK1325756) is a selective CXCR2 antagonist.
More description
|
|
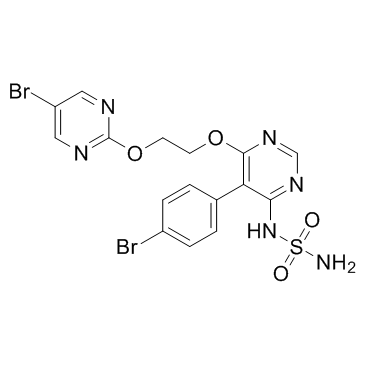
| DC11513 | Aprocitentan (ACT-132577) Featured |
Aprocitentan (ACT-132577) is the major and pharmacologically active metabolite of macitentan, which is dual ETA/ETB antagonist designed for tissue targeting.
More description
|
|