 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
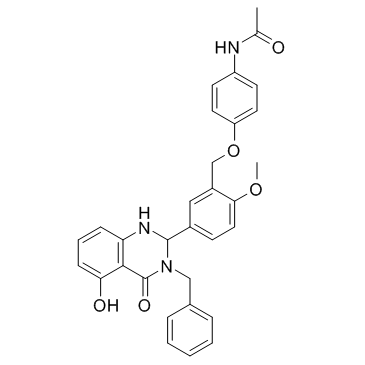
| DC12333 | ML-109 Featured |
ML-109 is a potent and full thyroid stimulating hormone receptor (TSHR) agonist, with an EC50 of 40 nM.
More description
|
|
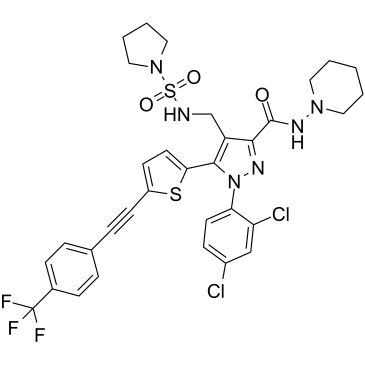
| DC7790 | CB1-IN-1(BPRCB1184) Featured |
CB1-IN-1 (BPRCB1184) is a peripherally restricted CB1R antagonist, with Ki of 0.3 nM and 21 nM for CB1R (EC50 = 3 nM) and CB2R, respectively. IC50 value: 0.3 nM (Ki, CB1R) 21 nM (Ki, CB2R) Target: CB1R in vivo: CB1-IN-1 is a novel peripherally restricted cannabinoid 1 receptor antagonist with significant weight-loss efficacy in diet-induced obese mice. CB1-IN-1 has great potential to ameliorate this related metabolic syndrome.
More description
|
|
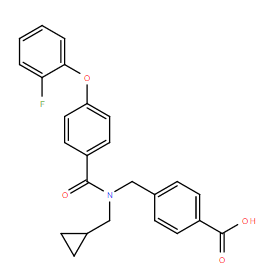
| DC10923 | TAK-615 Featured |
TAK-615 (TAK615) is a potent, selective, negative allosteric modulator (NAM) of the LPA1 receptor, partially inhibits the LPA response with IC50 of 91 nM (60% at 10 uM) in calcium mobilisation assays..
More description
|
|
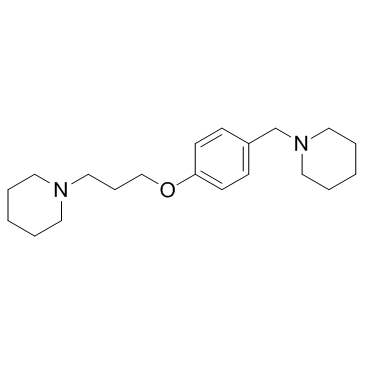
| DC12166 | JNJ-5207852 Featured |
JNJ-5207852 is a selective and potent histamine H3 receptor (H3R) antagonist, with pKis of 8.9, 9.24 for rat and human H3R, respectively.
More description
|
|
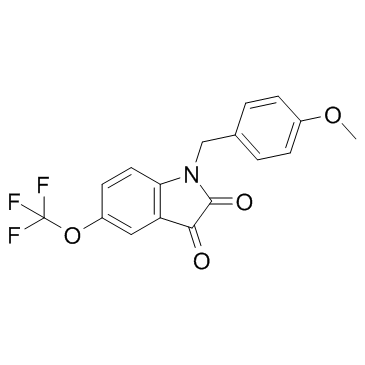
| DC12159 | VU 0238429 Featured |
VU 0238429 is positive allosteric modulator of muscarinic acetylcholine receptor subtype 5 (mAChR5 or M5), with an EC50 of 1.16 μM.
More description
|
|
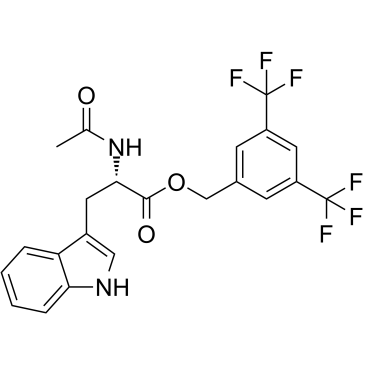
| DC11755 | L-732138 Featured |
A potent, selective and competitive NK1 receptor antagonist with IC50 of 2.3 nM.
More description
|
|
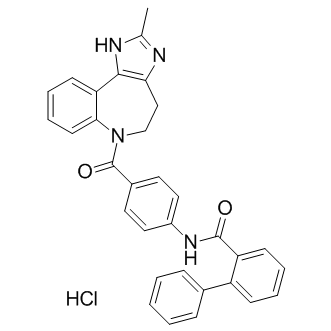
| DCAPI1091 | Conivaptan hydrochloride Featured |
Conivaptan(YM 087) is a non-peptide inhibitor of antidiuretic hormone (vasopressin receptor antagonist) with Ki values of 0.48 and 3.04 nM for rat liver V1A receptor and rat kidney V2 receptor respectively.
More description
|
|
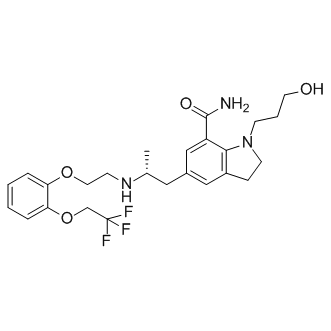
| DCAPI1485 | Silodosin Featured |
Silodosin(Rapaflo) is an α1-adrenoceptor antagonist with high uroselectivity. Silodosin causes practically no orthostatic hypotension (in contrast to other α1 blockers). Since Silodosin is a highly selective inhibitor of the α1A adrenergic receptor, it ca
More description
|
|
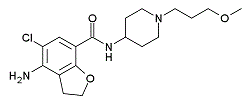
| DC4147 | Prucalopride Featured |
Prucalopride is a selective, high affinity 5-HT receptor agonistfor 5-HT4a and 5-HT4b with Ki of 2.5 nM and 8 nM, respectively.
More description
|
|
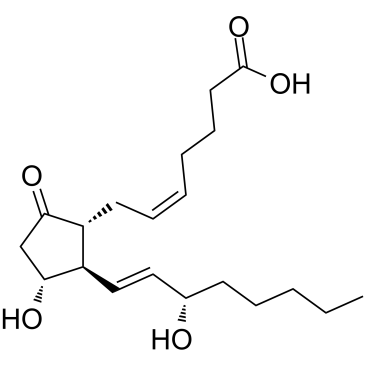
| DCAPI1163 | Prostaglandin E2 (Cervidil) Featured |
Prostaglandin E2 is a hormone-like substance that participate in a wide range of body functions such as the contraction and relaxation of smooth muscle, the dilation and constriction of blood vessels, control of blood pressure, and modulation of inflammation.
More description
|
|
| DC28780 | LY2444296 Featured |
LY2444296 is an orally bioavailable, high-affinity and selective short-acting kappa opioid receptor (KOPR) antagonist, with a Ki value of ∼1 nM. LY2444296 exhibits anti-anxiety like effects.
More description
|

|
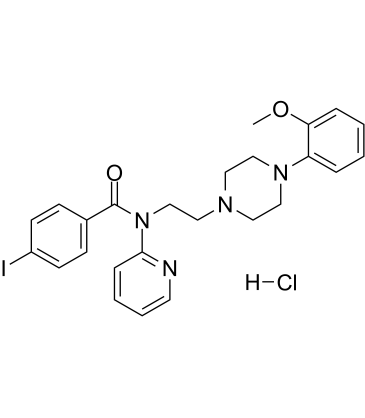
| DC28414 | p-MPPI hydrochloride Featured |
p-MPPI hydrochloride is a selective 5-HT1A receptor antagonist with high affinity for 5-HT1A receptors. p-MPPI hydrochloride can crosses the blood-brain barrier, and has clear antidepressant and anxiolytic-like effects.
More description
|
|
| DC10290 | PZM21 Featured |
PZM21 is a potent and selective μ opioid receptor agonist with an EC50 of 1.8 nM.
More description
|
|
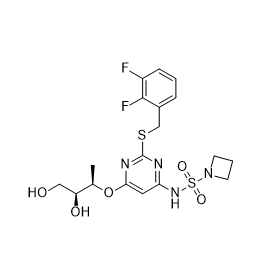
| DC10773 | AZD5069 Featured |
AZD-5069 is a potent and selective CXCR2 antagonist with the potential to inhibit neutrophil migration into the airways in patients with COPD.
More description
|
|
| DC10561 | GSK-2018682 Featured |
GSK-2018682 is a sphingosine 1 phosphate receptor (S1PR)-1 agonist potentially for the treatment of multiple sclerosis.
More description
|
|
| DC49392 | (±)-Muscarine chloride |
(±)-Muscarine chloride is the racemate of Muscarine chloride. Muscarine is a prototype muscarinic acetylcholine receptor agonist.
More description
|

|
| DC49366 | Heterobivalent ligand-1 |
Heterobivalent ligand-1 (compound 26) is a heterobivalent ligand for the Adenosine A 2A-dopamine D 2 receptor heteromer (KDB1 A2AR=2.1 nM, KDB1 D2R= 0.13 nM).
More description
|
|
| DC49365 | Zendusortide |
Zendusortide is a sortilin binding peptide.
More description
|
|
| DC49342 | A6770 |
A6770 is an orally active, potent sphingosine 1-phosphate (S1P) lyase (S1PL) inhibitor. A6770 is phosphorylated and the phosphorylated form directly inhibits S1P lyased.A6770, a potential key metabolite of THI, induces a [3H]dhS1P increase.
More description
|
|
| DC49332 | (R)-(-)-α-Methylhistamine dihydrochloride |
(R)-(-)-α-Methylhistamine dihydrochloride is a potent, selective and brain-penetrant agonist of H3 histamine receptor, with a Kd of 50.3 nM. (R)-(-)-α-Methylhistamine dihydrochloride can enhance memory retention, attenuates memory impairment in rats.
More description
|
|
| DC49331 | 4-Methylhistamine dihydrochloride |
4-Methylhistamine (dihydrochloride) is the potent agonist of histamine 4 receptor (H4R). 4-Methylhistamine (dihydrochloride) has the potential for the research of immune-related diseases such as cancer and autoimmune disorders.
More description
|
|
| DC49315 | L-Glutamine-15N-1 |
L-Glutamine-15N-1 (L-Glutamic acid 5-amide-15N-1) is the 15N-labeled L-Glutamine. L-Glutamine (L-Glutamic acid 5-amide) is a non-essential amino acid present abundantly throughout the body and involved in many metabolic processes. L-Glutamine provides a source of carbons for oxidation in some cells.
More description
|
|
| DC49310 | L-Glutamine-15N2 |
L-Glutamine-15N2 (L-Glutamic acid 5-amide-15N2) is the 15N-labeled L-Glutamine. L-Glutamine (L-Glutamic acid 5-amide) is a non-essential amino acid present abundantly throughout the body and involved in many metabolic processes. L-Glutamine provides a source of carbons for oxidation in some cells.
More description
|
|
| DC49221 | L-Glutamine-5-13C |
L-Glutamine-5-13C (L-Glutamic acid 5-amide-5-13C) is the 13C-labeled L-Glutamine. L-Glutamine (L-Glutamic acid 5-amide) is a non-essential amino acid present abundantly throughout the body and involved in many metabolic processes. L-Glutamine provides a source of carbons for oxidation in some cells.
More description
|
|
| DC49220 | L-Glutamine-1-13C |
L-Glutamine-1-13C (L-Glutamic acid 5-amide-1-13C) is the 13C-labeled L-Glutamine. L-Glutamine (L-Glutamic acid 5-amide) is a non-essential amino acid present abundantly throughout the body and involved in many metabolic processes. L-Glutamine provides a source of carbons for oxidation in some cells.
More description
|
|
| DC49157 | GLP-1 receptor agonist 8 |
GLP-1 receptor agonist 8 is a potent agonist of GLP-1 R. GLP-1 receptor agonist 8 has the potential for the research of diabetes, obesity, and nonalcoholic fatty liver disease (NAFLD) (extracted from patent WO2019239319A1, compound 17).
More description
|
|
| DC49156 | Peptide YY (PYY) (3-36), Human |
Peptide YY (PYY) (3-36), Human is an endogenous appetite suppressing peptide. Peptide YY (PYY) (3-36), Human, a neuropeptide Y (NPY) Y2 receptor agonist, is a powerful inhibitor of intestinal secretion.
More description
|
|
| DC49133 | SSTR5 antagonist 2 hydrochloride |
SSTR5 antagonist 2 hydrochloride is a highly potent, oral active and selective somatostatin (receptor) subtype 5 (SSTR5) antagonist and has potential for the research of type 2 diabetes mellitus (T2DM).
More description
|
|
| DC49131 | GLP-1 receptor agonist 9 |
GLP-1 receptor agonist 9 is a GLP-1 receptor agonist, example 7, extracted from WO2020234726 A1.
More description
|
|
| DC49124 | GLP-1 receptor agonist 7 |
GLP-1 receptor agonist 7 is a potent agonist of glucagon-like peptide-1 (GLP-1). GLP-1 receptor agonist 7 has the potential for the research of GLP-1-associated diseases, disorders, and conditions including diabetes mellitus (extracted from patent WO2021219019A1, compound 130b).
More description
|
|