 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
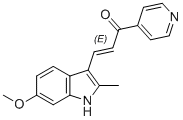
| DC12540 | 6-MOMIPP |
6-MOMIPP is a novel microtubule disruptor that targets the colchicine binding site on β-tubulin, induces mitotic arrest, caspase activation and loss of cell viability of U251 glioblastoma in vitro.
More description
|
|
| DC8864 | 6-Mercaptopurine (6-MP) Monohydrate |
6-Mercaptopurine Monohydrate is a widely used antileukemic agent and immunosuppressive drug that inhibits de novo purine synthesis through incorporation of thiopurine methyltransferase metabolites into DNA and RNA.
More description
|
 Monohydrate.gif)
|
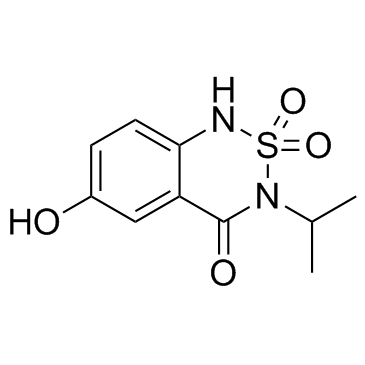
| DC12326 | 6-Hydroxybentazon (6-Hydroxybentazone) |
6-Hydroxybentazon is a phase I metabolite of bentazone, and bentazone is a chemical for use in herbicides.
More description
|
|
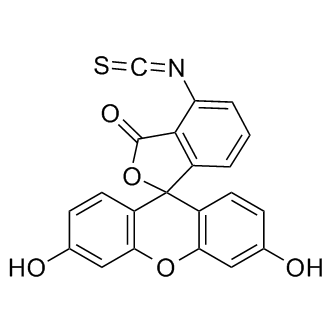
| DC9376 | 6-FITC |
6-Fluorescein isothiocyanate(6-FITC) is a derivative of fluorescein used in wide-ranging applications including flow cytometry.
More description
|
|
| DC20604 | 6-FABA |
6-FABA (6-Fluoroanthranilic Acid) is a small-molecule inhibitor of MTB tryptophan synthesis that converts Mtb into a tryptophan auxotroph and restores the efficacy of a failed host defense.
More description
|

|
| DC23336 | 6-Ethylthioinosine |
6-Ethylthioinosine (6-ETI.
More description
|
|
| DC20195 | 6-(Dimethylamino)purine;N,N-Dimethyladenine |
6-Dimethylaminopurine is a serine threonine protein kinase inhibitor. It inhibits the germinal vesicle breakdown and the meiotic maturation of oocytes.
More description
|

|
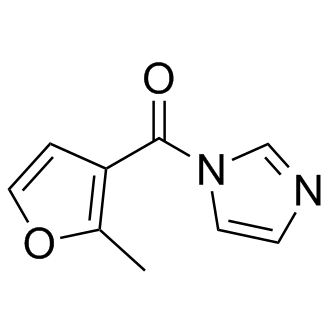
| DC7604 | FAI (5S rRNA modificator) Featured |
5S rRNA modificator is a suitable electrophile for 2’-hydroxyl acylation on structured RNA molecules, yielding accurate structural information comparable to that obtained with existing probes; 5S rRNA RNA modification.
More description
|
|
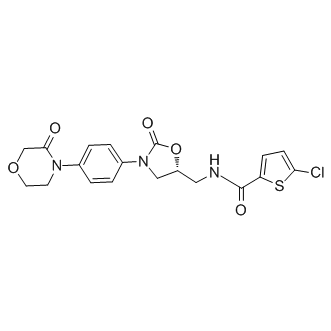
| DC9444 | 5-R-Rivaroxaban |
5-R-Rivaroxaban is (R) enantiomer of Rivaroxaban.
More description
|
|
| DC23334 | 5-NITP |
5-NITP (5-nitro-indolyl-2'-deoxyribose triphosphate) is a non-natural nucleotide that inhibits ribonucleotide reductase (hRR) with IC50 of 170 uM, demonstrates anti-cancer effects against leukemia cells by altering cell-cycle progression..
More description
|
|
| DC23335 | 5-NIdR |
5-NIdR (5-nitro-indolyl-2'-deoxynucleoside) is a non-natural nucleotide produces cytostatic and cytotoxic effects against human leukemia cells by altering cell-cycle progression.
More description
|
|
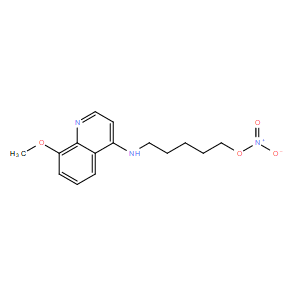
| DC10990 | 5MPN |
5MPN a first-in-class, selective, competitive, orally available inhibitor of PFKFB4 with Ki of 8.6 uM, does not inhibit PFK-1 or PFKFB3.
More description
|
|
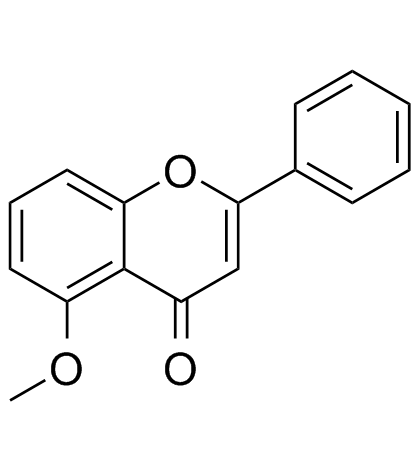
| DC20150 | 5-Methoxyflavone |
5-Methoxyflavone, belonged to Flavonoid family, is a DNA polymerase-beta inhibitor and neuroprotective agent against beta-amyloid toxicity. possess central nervous system (CNS) depressant effect mediated through the ionotropic GABAA receptors.
More description
|
|
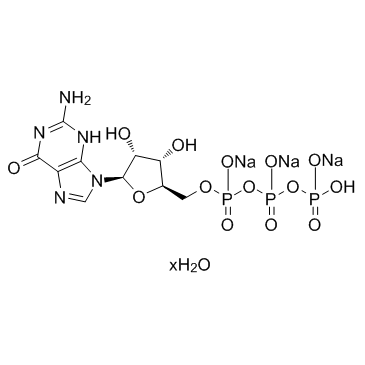
| DC12335 | 5'-GTP trisodium salt hydrate (Guanosine 5'-triphosphate trisodium salt hydrate) |
5'-GTP trisodium salt hydrate is an activator of the signal transducing G proteins and also serves as an energy-rich precursor of mononucleotide units in the enzymatic biosynthesis of DNA and RNA.
More description
|
|
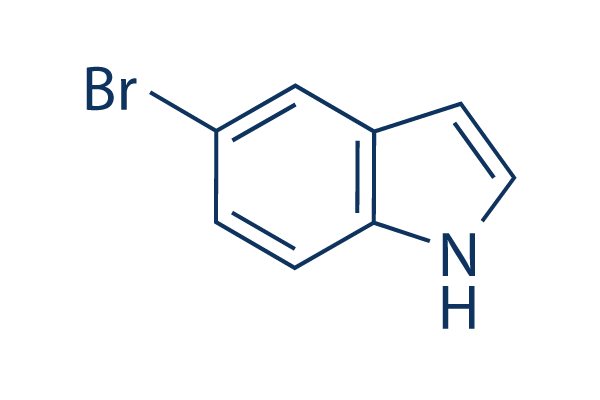
| DC20202 | 5-Bromoindole |
5-bromoindole is an important pharmaceutical chemical intermediate and a potential inhibitor of glycogen synthase kinase 3 (GSK-3).
More description
|
|
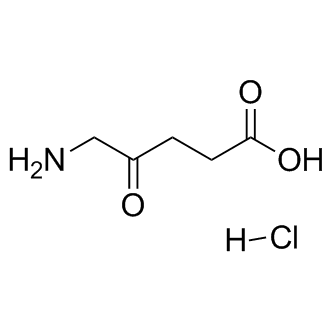
| DCAPI1240 | 5-Aminolevulinic acid HCl |
5-Aminolevulinic acid HCl
More description
|
|
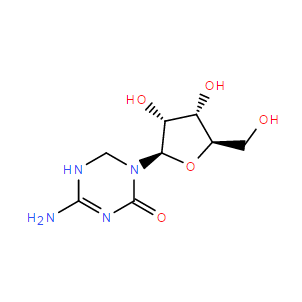
| DC9997 | 5,6-Dihydro-5-azacytidine |
5,6-Dihydro-5-azacytidine|cas 62488-57-7
More description
|
|
| DC20167 | 4-Hydroxyquinazoline;Quinazolin-4-ol, 4-Quinazolinol |
4-Hydroxyquinazoline is a PARP inhibitor with a high potency for PARP-1 and no effects on enzymes other than PARP.
More description
|

|
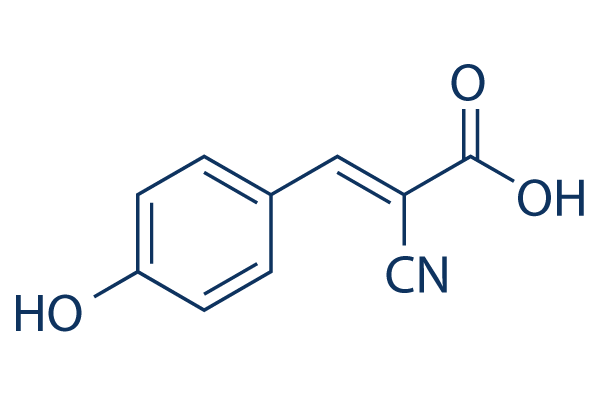
| DC10644 | α-CHCA |
4-Chloro-α-cyanocinnamic acid (α-CHCA) is a classic monocarboxylate transporters (MCT) inhibitor. α-cyano-4-hydroxycinnamate (CHC) has a 10-fold selectivity for MCT1 compared to other MCTs.
More description
|
|
| DC22400 | 4-BBPB maleate |
4-BBPB maleate is a highly potent agonist of σ1 receptor with Ki of 0.8 nM.
More description
|
|
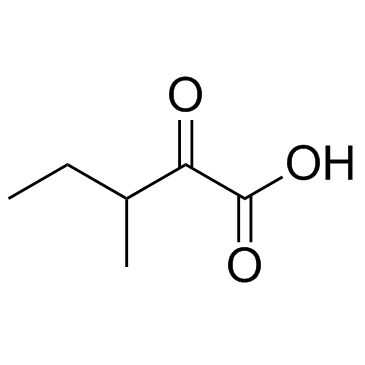
| DC12214 | 3-Methyl-2-oxovaleric acid |
3-Methyl-2-oxovaleric acid is a neurotoxin, an acidogen, and a metabotoxin, and also an abnormal metabolite that arises from the incomplete breakdown of branched-chain amino acids.
More description
|
|
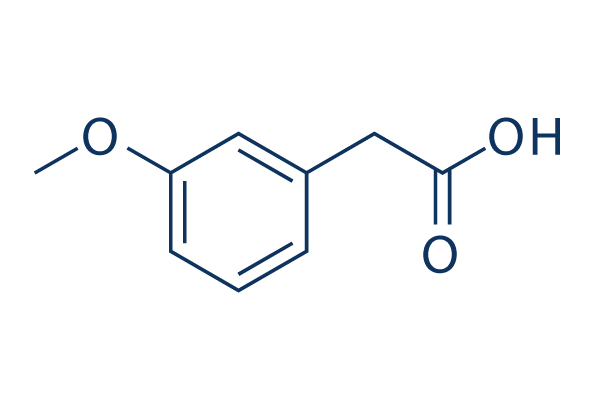
| DC20214 | 3-Methoxyphenylacetic acid;m-Methoxyphenylacetic acid, P-Methoxyphenylacetic acid, Anisylacetic acid, m-OMePAA |
3-Methoxyphenylacetic acid is a monocarboxylic acid.
More description
|
|
| DCAPI1054 | 3-Indolebutyric acid (IBA) |
3-Indolebutyric acid (IBA)
More description
|
|
| DC22994 | 3-Ethoxy-5,6-dibromosalicylaldehyde |
3-Ethoxy-5,6-dibromosalicylaldehyde is a potent and selective inhibitor of IRE1 endoribonuclease with IC50 of 0.12 uM.
More description
|
|
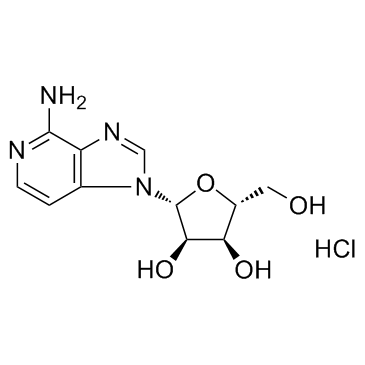
| DC12357 | 3-Deazaadenosine hydrochloride |
3-Deazaadenosine (hydrochloride) is an inhibitor of S-adenosylhomocysteine hydrolase, with a Ki of 3.9 µM; 3-Deazaadenosine has anti-inflammatory, anti-proliferative and anti-HIV activity.
More description
|
|
| DC20296 | 3-Cyanochromone |
3-Cyanochromone is a potent gram-negative bacteria WcbL protein inhibitor with IC50 of 28 uM in a competitive enzyme-inhibition model, shows inhibition constants Ki of 10 uM..
More description
|
|
| DC12240 | 3b-Hydroxy-5-cholenoic acid |
3b-Hydroxy-5-cholenoic acid is a monohydroxy bile acid of endogenous origin and could be found in children with the syndrome of hepatic ductular hypoplasia.
More description
|

|
| DC23038 | Acetylaconitine |
3-Acetylaconitine, aconitine, and deoxyaconitine are main toxic components of the roots of Aconitum pendulum.
More description
|

|
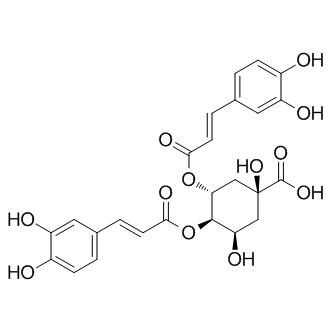
| DC23033 | Isochlorogenic acid B Featured |
3,4-Dicaffeoylquinic acid (3,4-DCQA) is a polyphenol with diverse biological activities.
More description
|
|
| DC8572 | 2-Phenyl-2-(1-piperidinyl)propane |
2-Phenyl-2-(1-piperidinyl)propane is an analog of phencyclidine that acts as a mechanism-based inactivator of human cytochrome P450 (CYP) 2B6 (Ki = 5.6 µM; IC50 = 5.1 µM).
More description
|
|