| DC70055 |
BTK-IN-9 |
BTK-IN-9 is a reversible BTK inhibitors with potent antiproliferative activity in mantle cell lymphoma. BTK-IN-9 specifically disturbs mitochondrial membrane potential and increases reactive oxygen species level in Z138 cells. BTK-IN-9 also induces cell apoptosis in Z138 cells. |

|
| DC70056 |
BLK-IN-2 |
BLK-IN-2 (compound 25) is a potent and selective irreversible inhibitor of B-Lymphoid tyrosine kinase (BLK), with an IC50 of 5.9 nM. BLK-IN-2 also inhibits BTK (IC50=202.0 nM). BLK-IN-2 shows potent antiproliferative activities against several lymphoma cells. |
|
| DC70057 |
BTK inhibitor 20 |
BTK inhibitor 20 is a potent BTK inhibitor with an IC50 of 8 nM. |
|
| DC70058 |
BTK-IN-8 |
BTK-IN-8 is a potent selective peripheral covalent BTK inhibitor (IC50=0.22 nM; Kd=0.91 nM). BTK-IN-8 has good whole blood CD69 cellular potency (IC50=0.029 µM). |
|
| DC70059 |
NSC381467 |
NSC381467 is a potent and orally active inhibitor of EGFR tyrosine kinase (EGFR-TK). NSC381467 has strong antiproliferative activities. NSC381467 has the potential for the research of cancer diseases. |
|
| DC70060 |
NSC114126 |
NSC114126 is a potent and orally active inhibitor of EGFR tyrosine kinase (EGFR-TK). NSC114126 has strong antiproliferative activities. NSC114126 has the potential for the research of cancer diseases. |
|
| DC70076 |
Pyrotinib |
Pyrotinib (SHR-1258) is a potent and selective EGFR/HER2 dual inhibitor with IC50 of 13/38 nM, respectively. |
|
| DC70083 |
PRN473 |
PRN473 (Atuzabrutinib, SAR 444727, PRN-473) is a potent, selective, covalent reversible BTK inhibitor. |
|
| DC70096 |
SRI-37264 |
SRI-37264 (SRI 37264) is a small molecule inhibitor of HIV-1 Nef-mediated activation of the myeloid Src-family kinase Hck, blocks HIV-1 replication in macrophages and disrupts MHC-I downregulation. |
|
| DC70102 |
ITK inhibitor
Featured
|
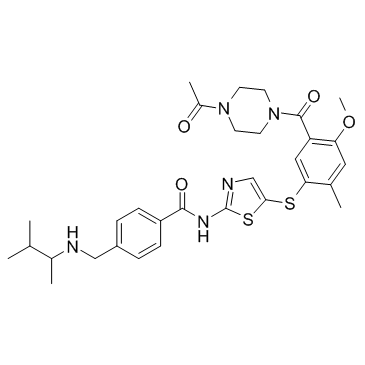
ITK inhibitor (CAS 439574-61-5 ) is a Interleukin-2-inducible T-cell kinase (Itk) inhibitor.. |
|
| DC70170 |
AC3573 |
AC3573 (AC-3573) is a potent, specific small molecule inhibitor of HER3.AC3573 abrogates HER2–HER3 signalling in cells and is more specific for HER3, inhibits NRG (heregulin)-induced HER3 phosphorylation with IC50 of 10 uM, abrogates the formation of the active HER2-HER3 heterodimer, inhibits oncogenic downstream signalling in SK-BR-3 breast cancer cells. |
|
| DC70179 |
AL906 |
AL906 (AL 906) is a novel potent, selective EGFR inhibitor with IC50 of 12 nM (EGFR phosphorylation).AL906 blocked EGF-induced EGFR phosphorylation at doses as low as 1 uM in EGF-stimulated OV90 and DU145 cells.AL906 growth inhibitory activity was superior to that of the clinical drug gefitinib.AL906 selectively inhibited the growth of the NIH3T3-EGFR and HER2 transfectants in isogenic NIH3T3 transfected cell panel. |
|
| DC70182 |
ALK2 R206H inhibitor 23 |
ALK2 R206H inhibitor 23 is a potent and selective inhibitor of Activin Receptor-Like Kinase-2 (ALK2/ACVR1) with IC50 of 8 nM for mutant ALK2 R206H.ALK2 R206H inhibitor 23 displays >15-fold selectivity over ALK1/ALK3, and >100-fold over ALK5/ALK6. |
|
| DC70185 |
Alphastatin-C |
Alphastatin-C is a 14-amino acids peptide consisting of a C-terminal fragment of the α-chain of Fgn, is a potent inhibitor of bFGF induced endothelial cell (HUVEC-CS) activation in vitro.Alphastatin-C inhibits tumor angiogenesis and reduces melanoma tumor growth, inhibits the chorioallantoic membrane (CAM) angiogenesis in chick model.Alphastatin-C efficiently reduces tumor number and volume in a melanoma mice model, due to the impairment of tumor neovascularization in treated mice.Alphastatin-C is an efficient new antiangiogenic FGF-associated agent in vitro, and inhibitor of embryonic and tumor vascularization in vivo, also is an arteriogenic agent |
|
| DC70214 |
ASK120067 |
ASK120067 is a novel third-generation inhibitor of EGFR T790M (L858R/T790M IC50=0.3 nM, T790M IC50=0.5 nM), with selectivity over EGFR WT (IC50=6 nM).ASK120067 potently inhibited the EGFR L858R/T790M and EGFR T790M resistant mutants, with IC50 of 0.3 nM and 0.5 nM, respectively, as well as the EGFR exon19del sensitizing mutant (IC50= 0.5 nM).ASK120067 also displayed a favorable selectivity profile against a panel of 258 kinases.ASK120067 selectively inhibits the growth of EGFR-mutant cell lines and induces apoptosis, with IC50 values of 12 nM, 6 nM and 2 nM against NCI-H1975, PC-9, and HCC827 cells, respectively.ASK120067 dose-dependently inhibited EGF-induced EGFR L858R/T790M phosphorylation and consequent activation of the downstream molecules AKT and ERK in NCI-H1975 cells, with similar or even more effective potency than osimertinib.ASK120067 (5, 10 mg/kg once daily) demonstrated profound and selective antitumor efficacy in EGFR-mutant xenograft models in vivo. |
|
| DC70232 |
AZD 1332 |
AZD1332 is a potent, selective, ATP-competitive neurotrophic tyrosine kinase receptor (NRTK;TRK) inhibitor with IC50 of <10 nM; inhibits phosphorylation of TrkA/B/C in MCF10A cells overexpressing TrkA and NIH3T3 cells over expressing TrkA, TrkB, or TrkC with IC50 of <5 nM, inhibits a panel Baf3 survival driven by TrkA, TrkB, or TrkC with EC50 of 2 nM, AZD 1332 is orally available. |
|
| DC70250 |
BI-4020 |
BI-4020 (BI 4020) is a potent, next generation, wt-sparing inhibitor of EGFR mutant T790M and/or C797S, and EGFRdel19 T790M C797S (0.2 nM).BI-4020 inhibits not only the triple mutant EGFRdel19 T790M C797S variant but also the double mutant EGFRdel19 T790M and primary mutant EGFRdel19 while sparing activity against EGFRwt.BI-4020 shows high potency on EGFR mutant cells, high kinome selectivity, and good DMPK properties.BI-4020 exhibites tumor regressions in the human PC-9 (EGFRdel19 T790M C797S) triple mutant NSCLC xenograft model in mice. |
|
| DC70256 |
BJG-03-025 |
BJG-03-025 is a potent, highly selective FAK tyrosine kinase inhibitor with biochemical IC50 of 20.2 nM.BJG-03-025 demonstrates excellent kinome selectivity was profiled by KINOMEscan screening, PLK1 proved to be a false positive with IC50 of 8.3 uM, 100-300-fold selective for FAK.BJG-03-025 displays antiproliferative activity in three-dimensional (3D)-breast and gastric cancer models and impacts cellular signaling and migration.BJG-03-025 decreased phosphorylated FAK (pFAK Y397) in gastric cancer CDH1–/–RHOAY42C/+ organoid model, decreased aberrant signaling and proliferation of gastric cancer organoids. |
|
| DC70273 |
BT173
Featured
|
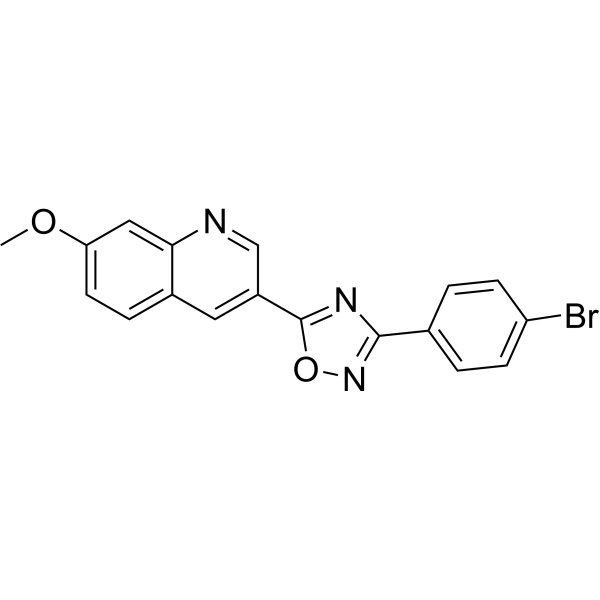
BT173 is a small molecule allosteric inhibitor of HIPK2-Smad3 interaction, specifically inhibits the TGF-β1/Smad3 pathway.BT173 disrupts HIPK2-Smad3 protein-protein interaction (PPI) without significant inhibition of HIPK2 kinase activity or inhibition of p53 activation.BT173 inhibits Smad3 phosphorylation in human kidney cells in vitro.BT173 significantly attenuated renal fibrosis development in the UUO mice, significantly decreased Smad3 phosphorylation and α-SMA expression in the UUO kidneys.Treatment of BT173 ameliorated kidney fibrosis in Tg26 mice. |
|
| DC70335 |
CXF-009 |
CXF-009 is a potent, specific, dual-warhead covalent inhibitor of FGFR4 (IC50=48 nM), forms dual-warhead covalent bonds with two cysteine residues in FGFR4.CXF-009 forms covalent bonds with FGFR4(C477A) and FGFR4(C552A), respectively; display weak inhibition against FGFR1-3 with IC50 of 2579 nM, 2408 nM, and 977 nM.CXF-009 shows inhibitory effect agaginst FGFR4 muntants FGFR4(C477A), FGFR4(C552A), and FGFR4(C477A, C552A) with IC50 of 193 nM, 602 nM, and 1528 nM, respectively.CXF-009 inhibited the growth of Ba/F3 cells transformed with FGFR1-4 with IC50 values of 1562 nM, 1971 nM, 777 nM, and 38 nM, respectively.CXF-009 is slightly more reactive to GSH than PRN1371. |
|
| DC70374 |
DYRK2 inhibitor C17
Featured
|
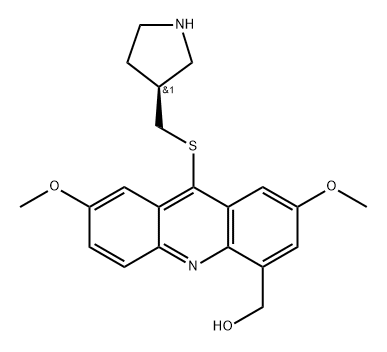
DYRK2 inhibitor C17 is a potent, selective DYRK2 inhibitor with IC50 of 9 nM, weakly inhibits DYRK3 (IC50=68 nM), and does not inhibit DYRK1A/1B (IC50>2000 nM).C17 showed outstanding selectivity for the human kinome containing 467 other human kinases.C17 showed synergistic 4E-BP1 phosphorylation suppression combined with AKT and MEK inhibitors.C17 impeded the store-operated calcium entry process (ORAI1 channel). |
|
| DC70383 |
EMI66 |
EMI66 (EMI-66) is a small molecule that attenuates RTK expression and signalling and alters the electrophoretic mobility of Coatomer Protein Complex Beta 2 (COPB2, binding Kd=1.51 uM) in lung cancer cells.EMI66 affects the subcellular localization of EGFR and COPB2, altering the endoplasmic reticulum (ER) stress response pathway, resulting in growth inhibition of mutant EGFR lung cancer cells and organoids.EMI66 inhibited total EGFR and MET levels, activation, and downstream signalling at lower concentrations than EMI1, also binds to recombinant COPB2 protein with an increased affinity compared to EMI1.EMI66 exerts an anti-proliferative effect (EC50=3-5 uM) on primary lung cancer organoid models, including XDO-137 (EGFR ex19del), PDXO-4000 (EGFR ex19del) and XDO-344 (wild-type EGFR). |
|
| DC70392 |
ER-001259851-000 |
ER-001259851-000 is a potent, selective, orally active Axl inhibitor with IC50 of 5.2 nM in cell free assays, displays >35-fold selectivity against Mer (IC50=190 nM).ER-001259851-000 shows a promising pharmacokinetic profile in mice. |
|
| DC70405 |
FF-10101 |
FF-10101 (FF10101) is a potent, selective, and irreversible FLT3 inhibitor with IC50 of 0.14 nM (FLT3 Wt), covalent binds to the C695 residue of FLT3.FF-10101 potently inhibited the phosphorylation of FLT3-ITD than that of FLT3-ITD-C695S (IC50 values 2.4 nM and 79 nM, respectively).FF-10101 potently and selectively inhibits the growth of mutant FLT3-expressing leukemia cells in vitro (MV4-11 c ell, IC50=0.83 nM), potently inhibits kinase activities of wild-type FLT3 and FLT3-D835Y with IC50 values of 0.20 nM and 0.16 nM, respectively.FF-10101 demonstrated high kinase selectivity to wild-type FLT3 with >30-fold margins against the other kinases except for FMS and KIT with IC50 values of 0.94 nM and 2.0 nM, respectively.FF-10101 showed growth inhibitory effects on all tested types of FLT3-TKD mutation- and FLT3-ITD with D835Y, Y842C, Y842H, or F691L mutation-expressing 32D cells with GI50 values from 0.43 nM to 6.1 nM.FF-10101 (2, 5, and 10 mg/kg) demonstrated anti-leukemic effects in in vivo models, FF-10101 more potently inhibited the growth of tumors with FLT3-ITD-D835Y and FLT3-ITD-F691L than quizartinib. |
|
| DC70408 |
FGFR4-IN-2 |
FGFR4-IN-2 is a potent, selective, covalent FGFR4 inhibitor with cellualr IC50 of 8.8 nM, 100-fold selectivity over FGFR2.FGFR4-IN-2 potently and selectively inhibit FGFR4 signaling through covalent modification of Cys552.FGFR4-IN-2 induces tumor regression in preclinical models of orthotopic and sorafenib-resistant HCC. |
|
| DC70447 |
GSK143 |
GSK143 is a highly potent, selective, orally efficacious Syk inhibitor with pIC50 of 7.5, >600-fold selective over ZAP-70; inhibits anti-IgM induced Erk1/2 phosphorylation in Ramos cells with pIC50 of 7.1, and anti-IgM induced CD69 surface expression in primary B cells with pIC50 of 6.6; shows good efficacy in the rat Arthus model. |
|
| DC70457 |
GSK2646264
Featured
|
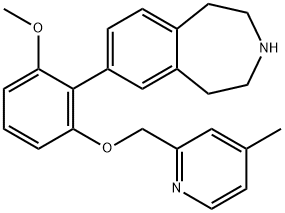
GSK2646264 (GSK-2646264) is a potent, selective Spleen Tyrosine Kinase (Syk) inhibitor with pIC50 of 7.1, >300-fold selectivity over Autota B; inhibits anti-IgM induced Erk1/2 phosphorylation in Ramos B cells with pIC50 of 6.7. |
|
| DC70488 |
HJ-01 |
HJ-01 is a small molecule that disrupts PTPσ-Trk interaction, enhances Trk signaling, and promotes sympathetic nerve regeneration over chondroitin sulfate proteoglycans (CSPGs). |
|
| DC70519 |
JBJ-08-178-01 |
JBJ-08-178-01 is a potent, selective and covalent HER2 inhibitor with IC50 of 2.21 nM and 7.04 nM for HER2 WT and EGFR WT, targets multiple HER2 activating mutations, including exon 20 insertions as well as amplification (IC50 values<50 nM).JBJ-08-178-01 maintained potency against non-insertion mutations, including S310F, L755S, V777L, V842I, and exon 19 indel (755_757LREdelinsRP) of HER2.JBJ-08-178-01 exhibited a much greater degree of HER2-selectivity in Ba/F3 cell lines than in biochemical assays, owing to weak growth inhibition against Ba/F3 cells with WT EGFR (IC50=368 nM).JBJ-08-178-01 showed strong antitumoral activity in HER2- mutant or amplified cancers in vitro and in vivo.Treatment with JBJ-08-178-01 also led to a reduction in total HER2 by promoting proteasomal degradation of the receptor.JBJ-08-178-01 is a selective, dual-action inhibitor and destabilizer of HER2 with potential efficacy and tolerance against NSCLC harboring HER2 genetic alterations or amplification. |
|
| DC70520 |
JBJ-09-063
Featured
|
JBJ-09-063 is a potent, mutant-selective allosteric EGFR inhibitor against EGFR L858R, T790M and C797S mutations, with IC50 of <0.1 nM for recombinant EGFR L858R/T790M kinase domain. |
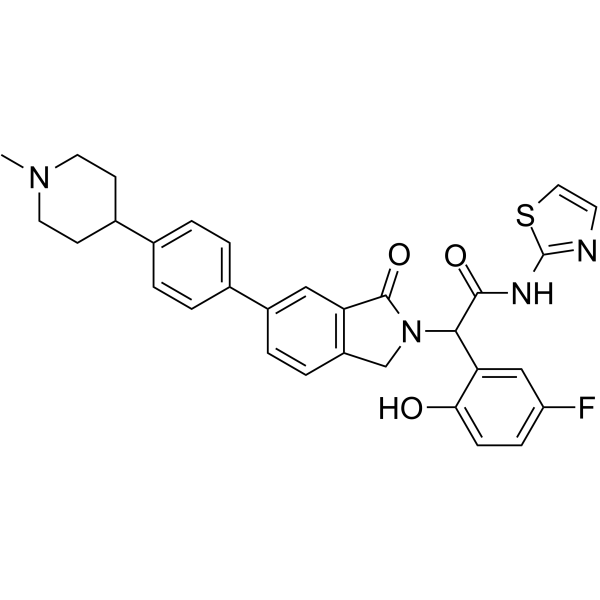
|
 DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.