 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC1080 | Tyrphostin 9(AG-17) Featured |
AG-17 is an inhibitor of epidermal growth factor (EGF) receptor kinase with an IC50 value of 460 µM in the human epidermoid carcinoma cell line A431.
More description
|

|
| DC12382 | AG-99 Featured |
AG-99 is an inhibitor of EGF receptor kinase with an IC50 value of 10 µM in the human epidermoid carcinoma cell line A431.
More description
|

|
| DC8072 | Altiratinib(DCC-2701) Featured |
Altiratinib is a MET/TIE2/VEGFR2/TRK (A,B,C) kinase inhibitor in Phase 1 clinical development for the treatment of invasive solid tumors including glioblastoma.
More description
|

|
| DC8845 | AMG337 Featured |
AMG-337 is a potent and highly selective small molecule ATP-competitive MET kinase inhibitor. AMG 337 inhibits MET kinase activity with an IC50 of < 5nM in enzymatic assays.
More description
|

|
| DC7324 | Amuvatinib (MP-470) Featured |
Amuvatinib (MP-470) is a potent and multi-targeted inhibitor of c-Kit, PDGFRα and Flt3 with IC50 of 10 nM, 40 nM and 81 nM, respectively.
More description
|

|
| DC10009 | Avitinib free base Featured |
Avitinib is an orally available, irreversible, epidermal growth factor receptor (EGFR) mutant-selective inhibitor, with potential antineoplastic activity.
More description
|

|
| DC10008 | Avitinib maleate Featured |
Avitinib is an orally available, irreversible, epidermal growth factor receptor (EGFR) mutant-selective inhibitor, with potential antineoplastic activity.
More description
|

|
| DC7746 | AZ191 Featured |
AZ191 is a potent and selective DYRK1B inhibitor with IC50 of 17 nM, about 5- and 110-fold selectivity over DYRK1A and DYRK2, respectively.
More description
|

|
| DC8462 | AZD-9291 mesylate (Osimertinib,Mereletinib) Featured |
AZD-9291 is a potent and selective mutated forms EGFR inhibitor(Exon 19 deletion EGFR IC50=12.92 nM, L858R/T790M EGFR IC50= 11.44 nM, wild type EGFR IC50= 493.8 nM).
More description
|

|
| DC10660 | BAY 61-3606 free base Featured |
BAY 61-3606 is a potent, ATP-competitive, reversible, and highly selective inhibitor of Syk tyrosine kinase activity (Ki= 7.5 nM) with no inhibitory effect against Btk, Fyn, Itk, Lyn, and Src.
More description
|

|
| DC9956 | BFH772 Featured |
BFH772 is a potent oral VEGFR2 inhibitor, which is highly effective at targeting VEGFR2 kinase with an IC50 value of 3 nM.
More description
|

|
| DC11819 | BMS-986142 Featured |
BMS-986142 is a potent, selective, and reversible BTK (Bruton’s tyrosine kinase) inhibitor (BTK IC₅₀ = 0.5nM; human WB IC₅₀ = 90 nM).
More description
|

|
| DC2019 | Brivanib (bms-540215) Featured |
Brivanib is an ATP-competitive inhibitor against human VEGFR2 and FGFR with IC50 of 25 nM and 148 nM, respectively.
More description
|

|
| DC48380 | Iruplinalkib |
Iruplinalkib (WX-0593) is a potent, selective, and orally active inhibitor of ALK and ROS1 tyrosine kinase. Iruplinalkib (WX-0593) shows favorable safety and promising antitumor activity in advanced NSCLC with ALK or ROS1 rearrangement.
More description
|

|
| DC48379 | Zilurgisertib |
Zilurgisertib is a selective ALK 2 inhibitor for treating diseases such as cancer.
More description
|
|
| DC48362 | OI338 |
OI338 is an orally available, ultralong-acting insulin analogue.
More description
|
|
| DC48361 | Dosimertinib |
Dosimertinib is a highly potent, selective, and orally efficacious deuterated EGFR targeting clinical candidate for the treatment of non-small-cell lung cancer.
More description
|
|
| DC48358 | BMS-986143 |
BMS-986143 is an orally active, reversible BTK inhibitor with an IC50 of 0.26 nM. BMS-986143 also inhibits TEC, BLK, BMX, TXK FGR, YES1, ITK with IC50s of 3 nM, 5 nM, 7 nM, 10 nM, 15 nM,19 nM, 21 nM, respectively. BMS-986143 can be used for the research of autoimmune diseases.
More description
|
|
| DC48357 | BCPyr |
BCPyr is a new candidate BTK degrader (DC50 = 800 nM).
More description
|
|
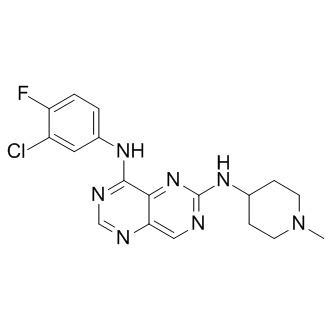
| DC7622 | BIBX1382 Featured |
BIBX 1382 is a potent, selective inhibitor of EGFR tyrosine kinase (IC50 = 3 nM); displays > 1000-fold lower potency against ErbB2 (IC50 = 3.4 μM) and a range of other related tyrosine kinases (IC50 > 10 μM).
More description
|
|
| DC8138 | CEP-28122 Featured |
CEP-28122 is a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers.
More description
|

|
| DC8009 | CEP-37440 Featured |
CEP-37440 is a novel potent and selective Dual FAK/ALK inhibitor with IC50 s of 2.3 nM (FAK) and 120 nM(ALK cellular IC50 in 75% human plasma).
More description
|

|
| DC7380 | CGI-1746 Featured |
CGI1746 is a small-molecule Btk inhibitor chemotype with a new binding mode that stabilizes an inactive nonphosphorylated enzyme conformation.
More description
|

|
| DC8728 | CH5424802(Alectinib HCl) Featured |
CH5424802(AF 802; Alectinib) is a potent ALK inhibitor with IC50 of 1.9 nM, sensitive to L1196M mutation.
More description
|

|
| DC10467 | CHMFL-BMX-078 Featured |
CHMFL-BMX-078 is a highly potent and selective type II irreversible BMX kinase inhibitor with an IC50 of 11 nM.
More description
|

|
| DC8651 | CO-1686 hydrobromide Featured |
CO-1686 hydrobromide is a novel, irreversible and orally delivered kinase inhibitor that specifically targets the mutant forms of EGFR including T790M (IC50=21 nM).
More description
|

|
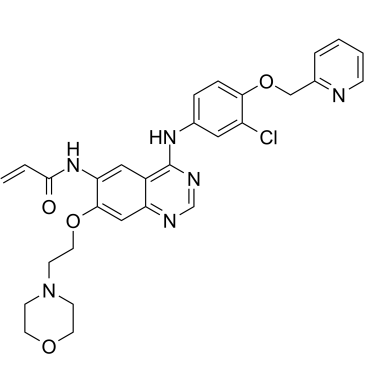
| DC41015 | BDTX-189 Featured |
BDTX-189 is a potent, orally active and selective inhibitor of allosteric EGFR and HER2 oncogenic mutations, includes EGFR/HER2 exon 20 insertion mutants. BDTX-189 shows KDs of 0.2, 0.76, 13 and 1.2 nM for EGFR, HER2, BLK and RIPK2, reapectively. Anticancer activity.
More description
|
|
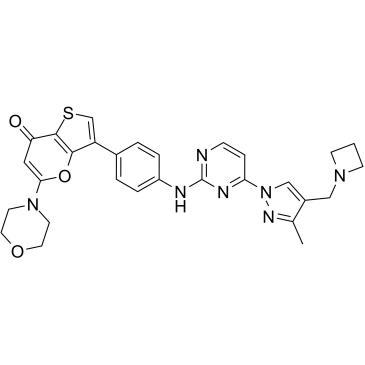
| DC40848 | SRX3207 Featured |
SRX3207 is an orally active and first-in-class dual Syk/PI3K inhibitor. SRX3207 possesses anti-tumor activity.
More description
|
|
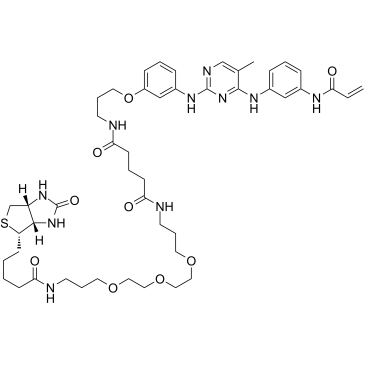
| DC28073 | CNX-500 Featured |
CNX-500 is a probe consisting of a covalent Btk inhibitor (CC-292) chemically linked to biotin. CNX-500 retains inhibitory activity against Btk (IC50 of 0.5 nM) and the ability to form a covalent bond with Btk. CNX-500 has low inhibitory effects on kinase epidermal growth factor receptor, and upstream Src-family kinases including Syk and Lyn.
More description
|
|
| DC44219 | FLT3-IN-3 Featured |
FLT3-IN-3 is a potent FLT3 inhibitor with IC50s of 13 and 8 nM for FLT3 WT and FLT3 D835Y, respectively.
More description
|
|