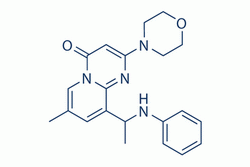
| Description: |
TGX-221 is a potent, selective, and cell membrane permeable inhibitor of the PI3K p110β catalytic subunit, used for cancer treatment. |
| Target: |
p110β:8.5 nM (IC50)
p110δ:211 nM (IC50) |
| In Vivo: |
TGX-221 (TGX221, 2.5 mg/kg i.v.) abolishes cyclic flow reductions in a Folts-like carotid artery stenosis preparation of thrombosis, without changing bleeding time, heart rate, blood pressure or carotid vascular conductance[4]. |
| In Vitro: |
TGX-221, BL05 and BL05-HA show selective cytotoxicity to LNCaP cells, which may be due to the deficiency of PTEN in this cell line and the accumulation of PIP3 in the cells[1]. TGX-221 (1 μM) does not affect the expression and phosphorylation of AMPK in C2C12 myoblasts[2]. TGX221 (0.1, 1, 10 µM) induces IL-6 release from ASM cells[2]. TGX-221 does not affect neurotensin-stimulated Akt phosphorylation when used alone, but it further suppresses neurotensin-stimulated phosphorylation of Akt when combined with gefitinib. TGX-221 abolishes the neurotensin-stimulated phosphorylation of Akt in Panc-1 cells[3]. |
| Cell Assay: |
The prostate cancer cell lines DU145 and LNCaP are maintained in RPMI-1640 medium, and PC3 cells are maintained in F-12K medium. LNCaP is a PSMA positive cell line, whereas DU145 and PC3 are PSMA negative. Both are supplemented with 10 % fetal bovine serum. Cells are plated in 96-well flat-bottomed plates at a concentration of 5,000 cells per well in 90 μL of growth medium. After 12 h, TGX-221, BL05, or BL05-HA loaded micelles in PBS are added at concentrations of 0, 0.1, 1, 5, 10, 50 or 100 μM. PBS and 10 μL of trichloroacetic acid (TCA) are added to negative and positive control wells, respectively. After 72 h, 10 μL of 55-μM resazurin blue is added to each well and incubated at 37°C for 4 h. After incubation, the resorufin product is measured with a fluorophotometer using an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The IC50 is determined as the midpoint between positive and negative control groups for each plate using GraphPad Prism 5 software. |
| Animal Administration: |
Rats are randomLy assigned to drug treatment groups consisting of the vehicle propylene glycol (0.25 mL/kg), LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; a reversible non-specific PI3K inhibitor; 10 mg/kg), wortmannin (an irreversible non-specific PI3K inhibitor; 5 mg/kg), IC87114 (2-[(6-aminopurin- 9-yl)methyl]-5-methyl-3-(2-methylphenyl)quinazolin-4-one; a PI3K p110δ antagonist; 2.5 mg/kg) and the selective PI3K p110β antagonist TGX221 (2.5 mg/kg). In the tail bleeding experiments, rats are randomLy assigned to drug treatment groups consisting of LY294002 (10 mg/kg), IC87114 (2.5 mg/kg), wortmannin (5 mg/kg), TGX221 (2.5 or 25 mg/kg), heparin (100 U/kg), aspirin (2× 200 mg/kg p.o.)± heparin (100 U/kg), and aspirin (2× 200 mg/kg p.o.) combined with heparin (100 U/kg) and TGX221 (2.5 mg/kg). All drugs, with the exception of aspirin, are administered as a slow (over ≈45-60 s) i.v. bolus of 0.25 mL/kg into the jugular vein. Aspirin (200 mg/kg suspended in 15% gum arabic in water) is administered twice orally (p.o.)-the first dose is given 24 h before the experiment and the second dose 1 h before the start of the experiment. |
| References: |
[1]. Zhao Y, et al. Prodrug strategy for PSMA-targeted delivery of TGX-221 to prostate cancer cells. Mol Pharm. 2012 Jun 4;9(6):1705-16.
[2]. Ge Q, et al. The phosphoinositide 3'-kinase p110δ modulates contractile protein production and IL-6 release in human airway smooth muscle. J Cell Physiol. 2012 Aug;227(8):3044-52.
[3]. Müller KM, et al. Role of protein kinase C and epidermal growth factor receptor signalling in growth stimulation by neurotensin in colon carcinoma cells. BMC Cancer. 2011 Oct 2;11:421.
[4]. Sturgeon SA, et al. Advantages of a selective beta-isoform phosphoinositide 3-kinase antagonist, an anti-thrombotic agent devoid of other cardiovascular actions in the rat. Eur J Pharmacol. 2008 Jun 10;587(1-3):209-15.
[5]. Chaussade C, et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J. 2007 Jun 15;404(3):449-58. |
 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.