 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
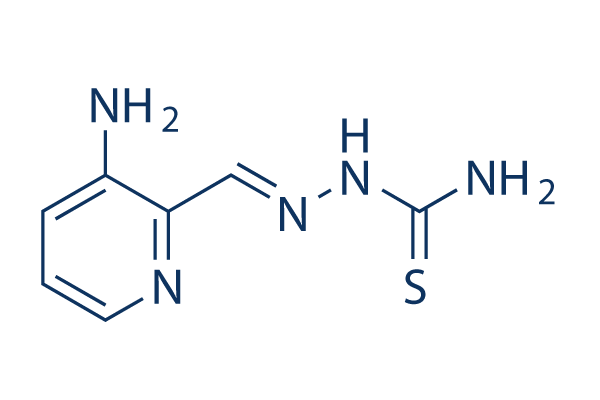




















| Cas No.: | 143621-35-6 |
| Chemical Name: | Hydrazinecarbothioamide, 2-[(3-amino-2-pyridinyl)methylene]-, (2E)- |
| Synonyms: | Triapine,NSC663249,3-AP,NSC 663249,NSC-663249,3AP,3 AP |
| SMILES: | N(C(N)=S)/N=C/C1=NC=CC=C1N |
| Formula: | C7H9N5S |
| M.Wt: | 195.24 |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | Triapine is a novel inhibitor of the M2 subunit of ribonucleotide reductase (RR), and is a potent radiosensitizer. |
| In Vivo: | Triapine causes a significant increase (1.7-fold) in splenic weight when expressed as a percentage of total body weight (1.02±0.06%; n=25) compared with control mice (0.6±0.03%; n=27). In the long-term group, a significant increase in heart weight is observed after Dp44mT (0.4 mg/kg per day) (0.8±0.06%; n=4) compared with control mice (0.5±0.01%; n=6). A significant decrease in the expression of Ndrg1, TfR1, and VEGF1 in the liver is noted for Dp44mT- and Triapine (12 mg/kg per day)-treated animals. The decreased expression could be related to the increased liver Fe in both Dp44mT- and Triapine-treated mice[3]. |
| In Vitro: | Triapine is a potent derivative of α-heterocyclic carboxaldehyde thiosemicarbazone (HCT) that inhibits hRRM2 and p53R2 isoforms of the M2 subunit[1]. Triapine is thought to inhibit ribonucleotide reductase through its preformed iron chelate, rather than directly by removing iron from the active site. In cells containing less topoisomerase IIα fewer DNA strand breaks will be produced, and thus topoisomerase II poisons will be less inhibitory in the K/VP.5 cell line. The IC50s for Dp44mT growth inhibition are 48±9 nM and 60±12 nM, for K562 and K/VP.5 cells, respectively. The IC50s for Triapine growth inhibition are 476±39 nM and 661±69 nM for K562 and K/VP.5 cells, respectively[2]. PKIH and DpT Fe chelators show high antiproliferative activity against a range of tumor cell lines. Dp44mT shows the greatest antitumor efficacy with an IC50 that ranged from 0.005 to 0.4 μM. The average IC50 of Dp44mT over 28 cell types is 0.03±0.01 μM, which is significantly lower than that of Triapine (average IC50: 1.41±0.37 μM)[3]. |