 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
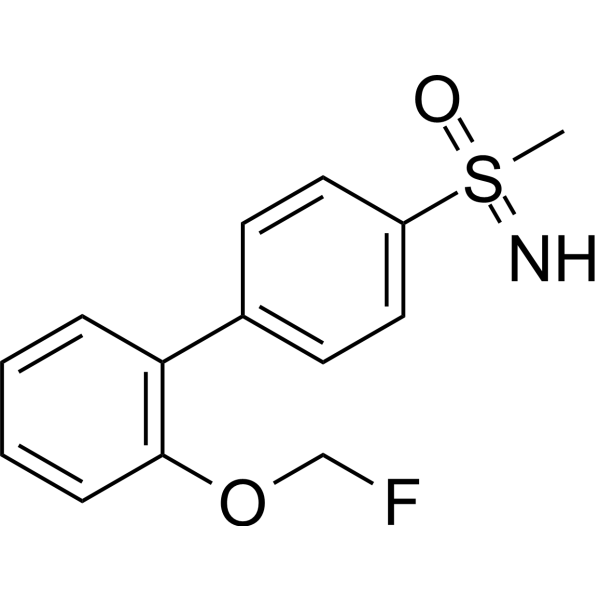
| DC72808 | UCM-1306 Featured |
UCM1306 is a potent and orally active human dopamine D1 receptor allosteric modulator (PAM). UCM-1306 increases the endogenous dopamine (DA) maximal effect both in human and mouse D1 receptors. UCM-1306 is not only for improving motor symptoms but also for addressing the key comorbid cognitive impairment associated with long-term Parkinson’s disease (PD).
More description
|
|
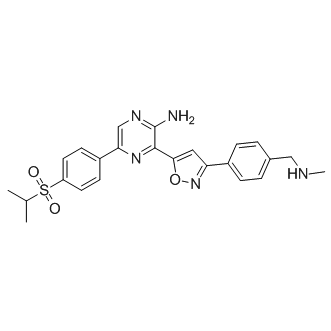
| DC7527 | VE-822 (Berzosertib) Featured |
VE-822 is a selective ATR inhibitor with an Ki value of 0.2 nM, >150 fold selectivity over ATM (Ki=34 nM), DNA-PK (Ki >4 uM) and mTOR (Ki >1 uM).
More description
|
|
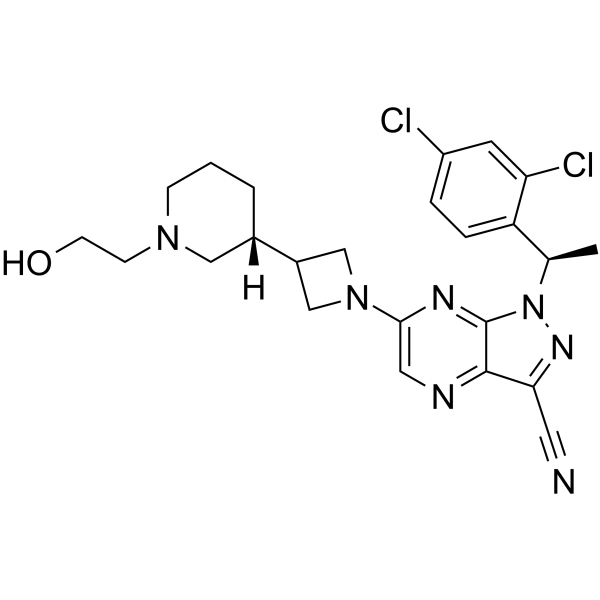
| DC70292 | CCR4-IN-38 Featured |
CCR4-IN-38 (CCR4-351) is a potent, selective, orally bioavailable CCR4 antagonist with IC50 of 50 nM (Chemotaxis inhibition).CCR4-IN-38 inhibits the recruitment of Treg into the tumor microenvironment (TME).CCR4-IN-38 elicits antitumor responses as a single agent or in combination with an immune checkpoint blockade.
More description
|
|
| DC65522 | Nico-52 Featured |
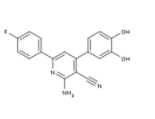
|
|
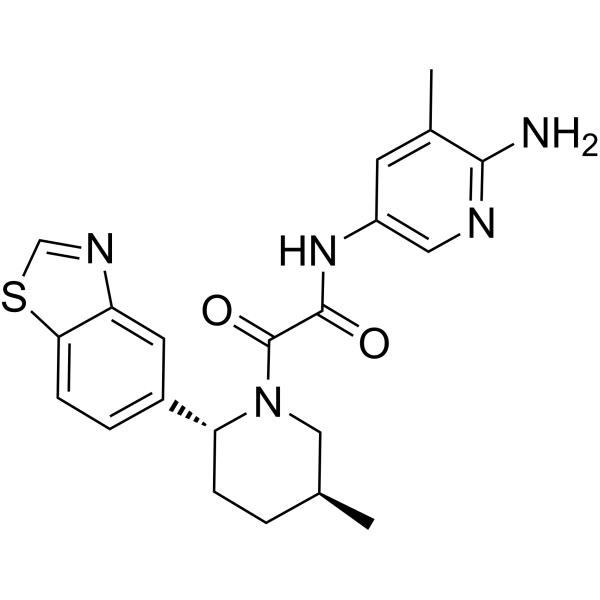
| DC65520 | TNG908 Featured |
TNG908 is a MTAP-cooperative PRMT5 inhibitor that can pass through the blood-brain barrier. TNG908 is 15 times more selective for MTAP null cell lines than MTAPWT cell lines, and can be used in cancer research.
More description
|
|
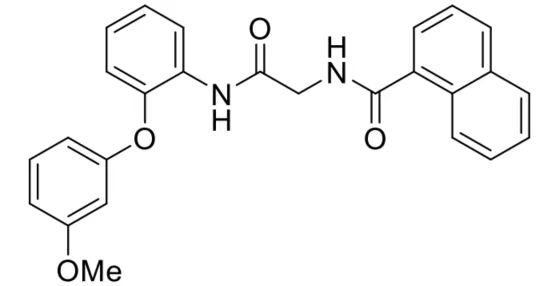
| DC60479 | AOH1996 Featured |
AOH1996 is a proliferating cell nuclear antigen (PCNA) inhibitor. AOH1996 enhances PCNA and RPB1 interaction and interferes with TRC resolution and induces DNA double-stranded breaks in a transcription dependent manner. AOH1996 almost completely inhibits the growth of xenograft tumors without causing any discernible toxicity to experimental animals.AOH1996 has superior metabolic stability as compared to the AOH1160 parent molecule.
More description
|
|
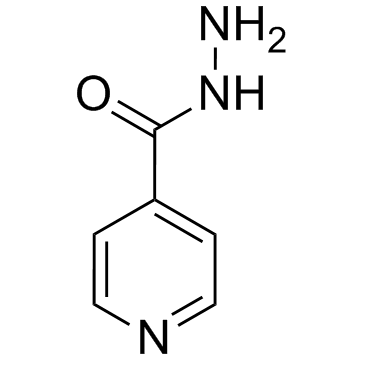
| DCAPI1242 | Isoniazid(Tubizid) Featured |
Isoniazid is an antibacterial agent used primarily as a tuberculostatic.Target: AntibacterialIsoniazid is a prodrug and must be activated by a bacterial catalase-peroxidase enzyme that in M. tuberculosis is called KatG [1]. KatG couples the isonicotinic acyl with NADH to form isonicotinic acyl-NADH complex. This complex binds tightly to the enoyl-acyl carrier protein reductase known as InhA, thereby blocking the natural enoyl-AcpM substrate and the action of fatty acid synthase. This process inhibits the synthesis of mycolic acid, required for the mycobacterial cell wall. A range of radicals are produced by KatG activation of isoniazid, including nitric oxide, which has also been shown to be important in the action of another antimycobacterial prodrug PA-824 [2, 3]. Isoniazid is bactericidal to rapidly dividing mycobacteria, but is bacteriostatic if the mycobacteria are slow-growing [4].
More description
|
|
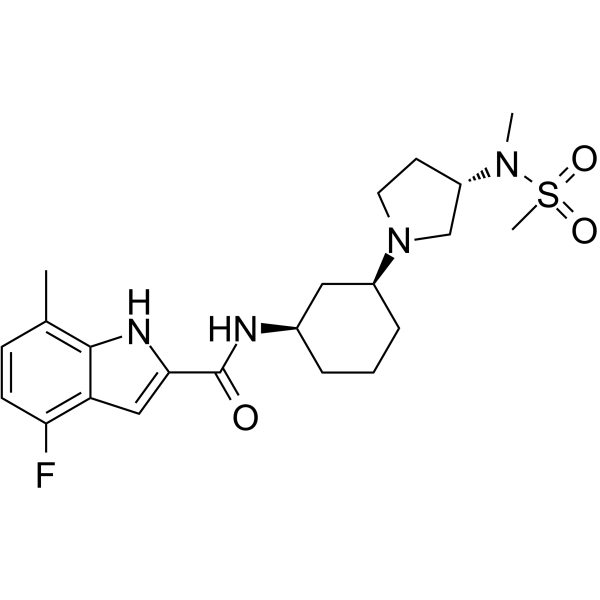
| DC47616 | EPZ-719 Featured |
EPZ-719 is a novel and potent SETD2 inhibitor ( IC50 = 0.005 μM) with a high selectivity over other histone methyltransferases.
More description
|
|
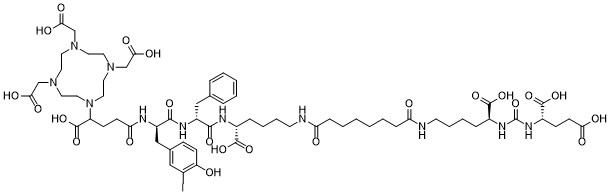
| DC65517 | PSMA-I&T Featured |
PSMA-I&T is a PSMA-based radioligand. [177Lu]Lu-PSMA I&T can be uused for potential treatment of metastatic castration-resistant prostate cancer. 67 Ga-PSMA I&T can be used for Radioguided Surgery of Lymph Node Metastases With Biochemical Recurrence of Prostate Cancer
More description
|
|
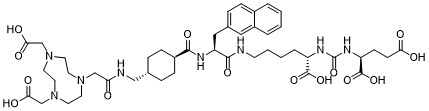
| DC65516 | PSMA-BCH Featured |
PSMA-BCH also known as NOTA-PSMA, is a PSMA- NOTA-conjugated precursor. 68Ga-PSMA-BCH can be used for prostate cancer imaging. Al18F-PSMA-BCH can be used for prostate cancer imaging as a novel 18F PET radiotracer. 64Cu-PSMA-BCH is a new radiotracer for delayed PET imaging of prostate cancer. 64Cu-PSMA-BCH was PSMA specific and showed high stability in vivo with lower uptake in liver than 64Cu-PSMA-617. Biodistribution in mice and PCa patients showed similar profile compared with other PSMA ligands and it was safe with moderate effective dosimetry.
More description
|
|
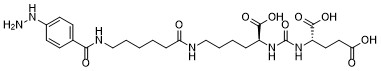
| DC65515 | HYNIC-PSMA Featured |
HYNIC-PSMA is a PSMA based ligand, which can bind radio-active metal ion and used as a tumor-imaging agent in prostate cancer. 99mTc-HYNIC-PSMA is a novel technetium-99m-labeled small-molecule inhibitor of prostate-specific membrane antigen (PSMA) for detection of prostate cancer. 99mTc-HYNIC-PSMA showed high tracer uptake (with a tumor-to-background ratio of 9.42 ± 2.62) in the malignant lesions of PCa patients, making it a promising radiopharmaceutical imaging method for site-specific management of PCa.
More description
|
|
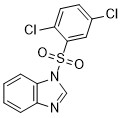
| DC65512 | WDR5-IN-6 Featured |
WDR5-IN-6 is a WDR5 inhibitor.
More description
|
|
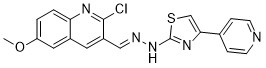
| DC65511 | Antibacterial agent 133 Featured |
Antibacterial agent 133 (4l) is an antimicrobial agent
More description
|
|
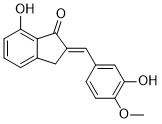
| DC65509 | TNF-α-IN-9 Featured |
TNF-α-IN-9 (compound 48) is a analog of NDM-1 inhibitor-3
More description
|
|
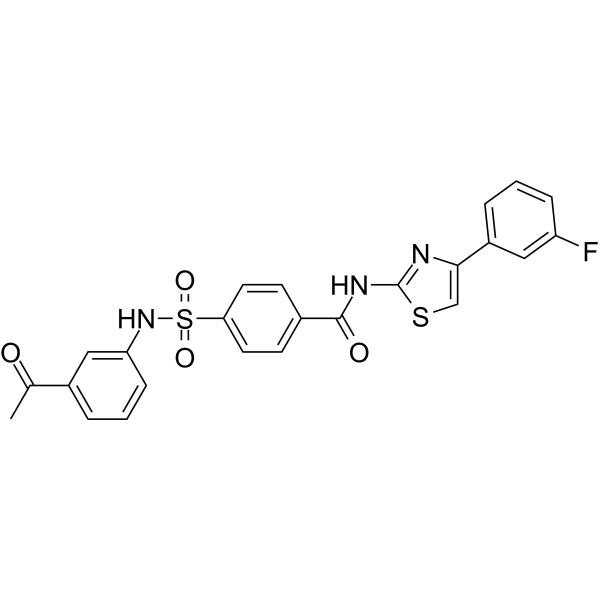
| DC65507 | PHGDH-IN-3 Featured |
PHGDH-IN-3 is an orally active phosphoglycerate dehydrogenase (PHGDH) inhibitor. PHGDH-IN-3 inhibits PHGDH with an IC50 value of 2.8 μM. PHGDH-IN-3 can be used for the research of cancer[1].
More description
|
|
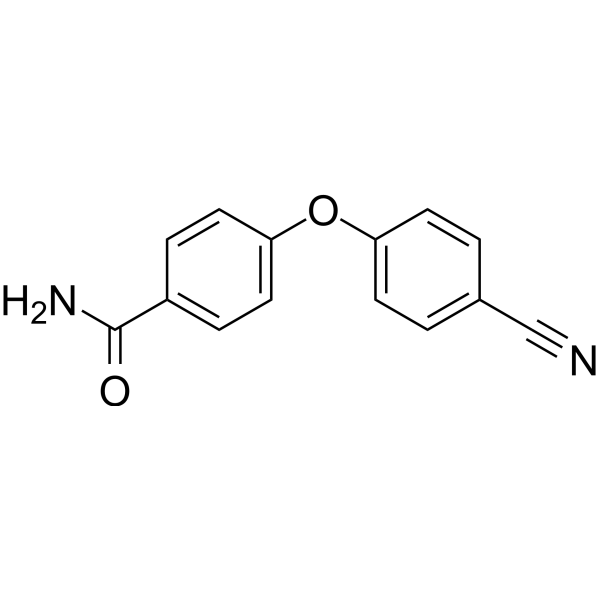
| DC65506 | PARP10-IN-2 Featured |
PARP10-IN-2 is a potent mono‐ADP‐ribosyltransferase PARP10 inhibitor with an IC50 of 3.64 μM for human PARP10. PARP10-IN-2 reveals potent inhibition on PARP2 and PARP15 with IC50s of 27 μM and 11 μM for human PARP2 and human PARP15, respectively[1].
More description
|
|
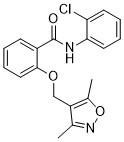
| DC72718 | S19-1035 Featured |
S19-1035 is a highly potent and specific aldo-keto reductase 1C3 (AKR1C3) inhibitor. S19-1035 inhibits AKR1C3 with an IC50 value of 3.04 nM. S19-1035 can be used for the research of tumor.
More description
|
|
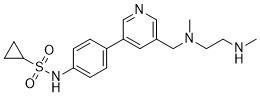
| DC65505 | PRMT6-IN-3 Featured |
PRMT6-IN-3 (compound 25) is a selective PRMT6 inhibitor
More description
|
|
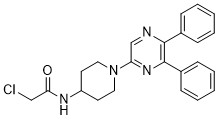
| DC65504 | Skp2 inhibitor 1 Featured |
Skp2 inhibitor 1 (compound 14i) is a potent and selective Skp2 inhibitor
More description
|
|
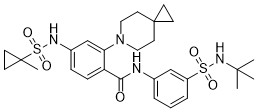
| DC65503 | KIF18A-IN-3 Featured |
KIF18A-IN-3 is a potent KIF18A inhibitor (IC50=61 nM). KIF18A-IN-3 causes significant mitotic arrest and increases the number of mitotic cells in tumor tissues. KIF18A-IN-3 can be used for researching cancer[1].
More description
|
|
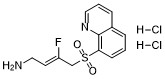
| DC47316 | LOX-IN-3 Dihydrochloride Featured |
LOX-IN-3 Dihydrochloride is an orally active and selective lysyl oxidase (LOX) inhibitor.
More description
|
|
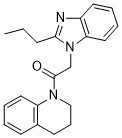
| DC65501 | GFP16 Featured |
GFP16 is a low affinity antiprion compound.
More description
|
|
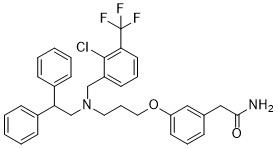
| DC65500 | GW6340 Featured |
GW6340 is an intestinal-specific LXR agonist.
More description
|
|
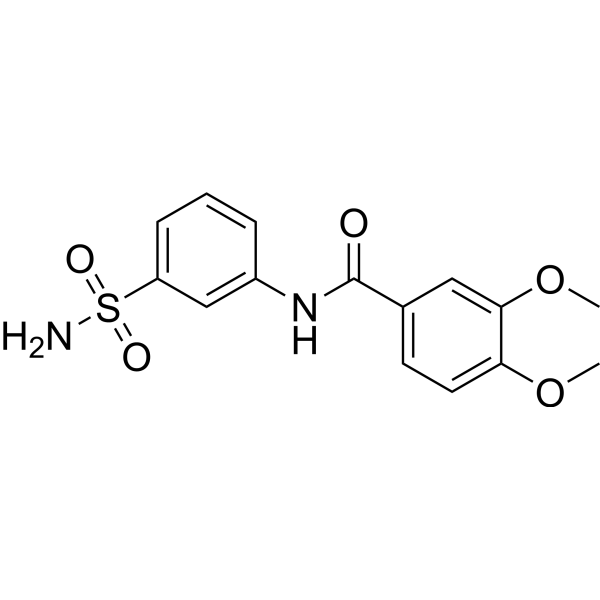
| DC65499 | hCAII-IN-8 Featured |
hCAII-IN-8, an amide, is a highly selective carbonic anhydrase (CA) inhibitor with an IC50 value of 0.18 μM against hCA II[1].
More description
|
|
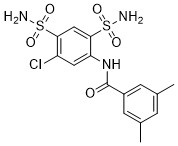
| DC65498 | hCAII-IN-9 Featured |
hCAII-IN-9 is a potent carbonic anhydrase inhibitor
More description
|
|
| DC65497 | CYP4Z1-IN-1 Featured |
CYP4Z1-IN-1 (compound 7c) is a potent CYP4Z1 inhibitor, with an IC50 of 41.8 nM. CYP4Z1-IN-1 decreases the expression of breast CSCs stemness markers, spheroid formation, and metastatic ability as well as tumor-initiation capability in a concentration-dependent manner in vitro and in vivo[1].
More description
|

|
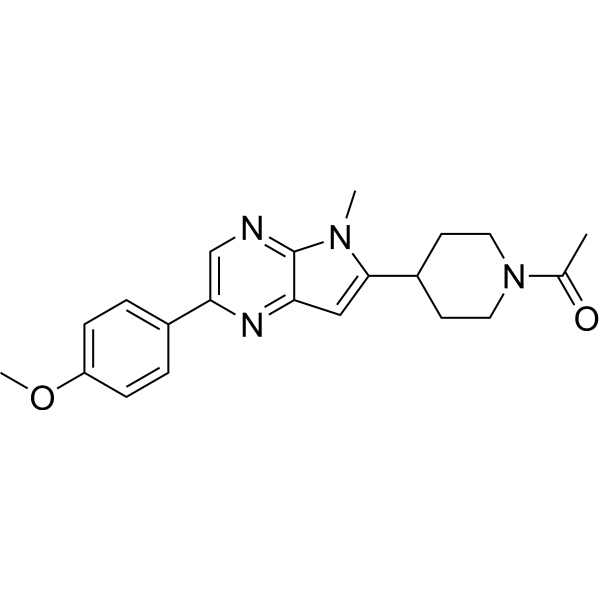
| DC65496 | Gcase activator 2 Featured |
Gcase activator 2 (compound 14), a pyrrolo[2,3-b]pyrazine, is alos a β-Glucocerebrosidase (GCase) activator (EC50=3.8 μM). Gcase activator 2 induces GCase dimerizatio (both K-type and V-type). And Gcase activator 2 has low metabolic clearance in human and mouse[1].
More description
|
|
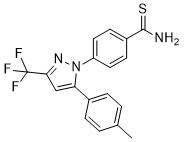
| DC65495 | PYZ18 Featured |
PYZ18 is a selective COX-2 inhibitor with an IC50 of 7.07 μM. PYZ18 is the best lead compound for COX-2 inhibitors. PYZ18 has anti-inflammatory and other biological properties[1].
More description
|
|
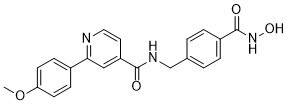
| DC65494 | HDAC-IN-57 Featured |
HDAC-IN-57 is an orally active inhibitor of histone deacetylases (HDAC), with IC50s of 2.07 nM, 4.71 nM, 2.4 nM and 107 nM for HDAC1, HDAC2, HDAC6, HDAC8, respectively. HDAC-IN-57 can inhibits LSD1, with IC50 of 1.34 μΜ. HDAC-IN-57 induces apoptosis, and has anti-tumor activity[1].
More description
|
|
| DC65493 | SIRT5 inhibitor 5 Featured |
SIRT5 inhibitor 5 is a potent SIRT5 inhibitor with an IC50 value of 0.21 µM.
More description
|
|