 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
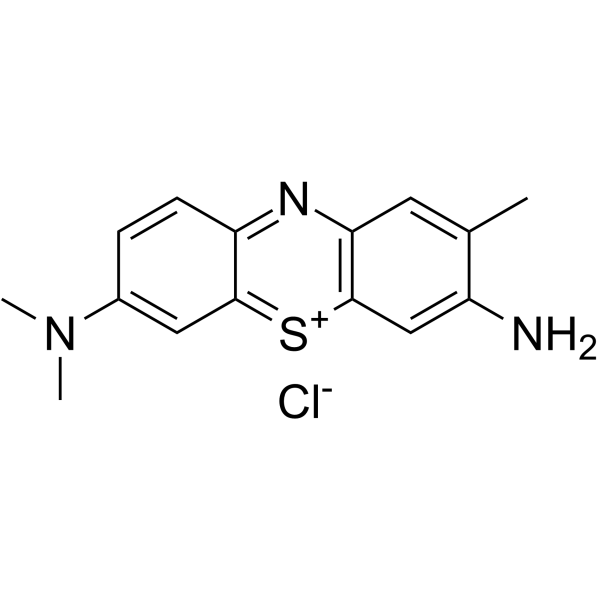
| DC48634 | Toluidine Blue Featured |
Toluidine Blue is a basic thiazine metachromatic dye with high affinity for acidic tissue components. Toluidine Blue has wide applications in vital staining in living tissues and a special stain.
More description
|
|
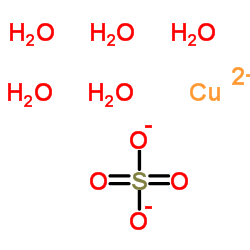
| DC65492 | Copper sulfate Featured |
Cupric sulfate (Copper(II) sulfate) is a salt formed by treating cupric oxide with sulfuric acid. Cupric sulfate is a lewis acid catalyst commonly used to promote acid-catalyzed organic transformations.
More description
|
|
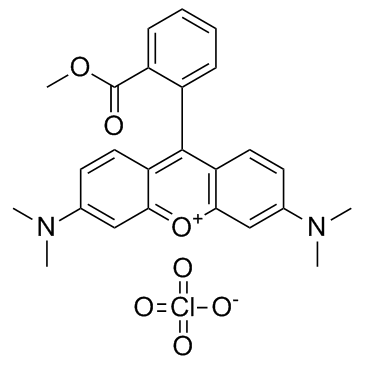
| DC65491 | TMRM Perchlorate Featured |
TMRM Perchlorate is a cell-permeant cationic lipophilic red fluorescent dye (λex=530 nm, λem=592 nm).
More description
|
|
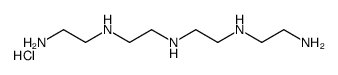
| DC65490 | Colestipol hydrochloride Featured |
Colestipol hydrochloride, a salt form of Colestipol, is a high-molecular-weight, insoluble, granular copolymer of tetraethylenepentamine and epichlorohydrin. It functions as an anion-exchange and resin-sequestering agent. Colestipol hydrochloride reduces cholesterol levels without affecting triglycerides.
More description
|
|
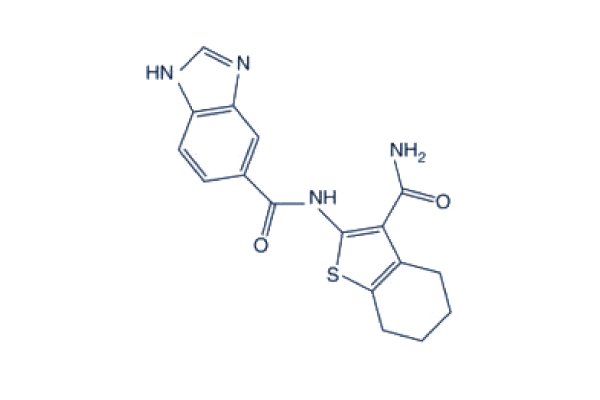
| DC65489 | CRCD2 Featured |
CRCD2 is an NT5C2 (cytosolic 5’ nucleotidase II) inhibitor with a kd of 70.9 μM. It enhances the cytotoxic effects of 6-MP and effectively reverses thiopurine resistance mediated by genetic and non-genetic mechanisms of NT5C2 activation in ALL.
More description
|
|
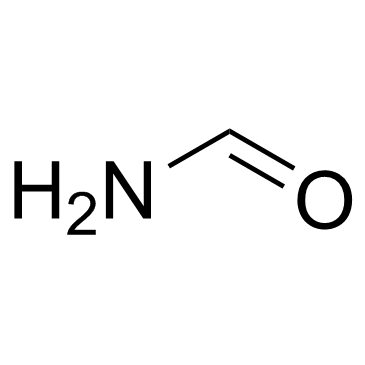
| DC72862 | Formamide Featured |
Formamide (Methanamide; Formimidic acid) is a clear liquid amide derived from formic acid. It allows for the denaturation and renaturation of nucleic acids at room temperature. Formamide-induced DNA denaturation when combined with detection of denatured DNA with a monoclonal antibody (MAb) makes it possible to specifically identify the apoptotic cells.
More description
|
|
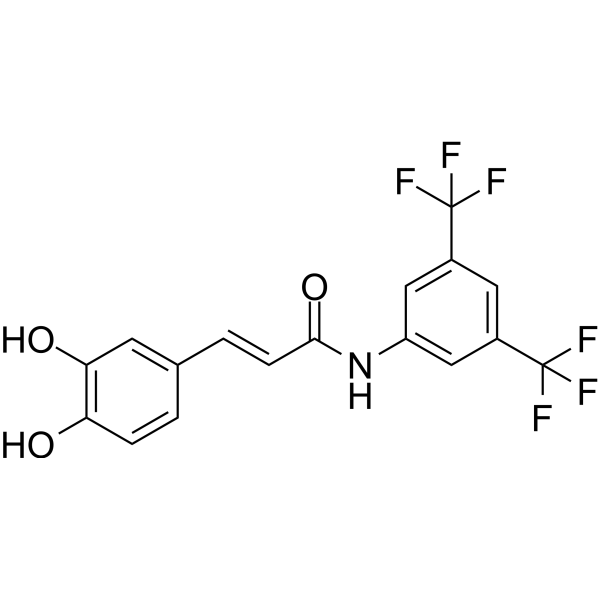
| DC65487 | SRD5A1-IN-1 Featured |
SRD5A1-IN-1 (Compound 4) is a competitive and covalent steroid 5α-reductase type 1 (SRD5A1) inhibitor with an IC50 of 1.44 µM. SRD5A1-IN-1 modulates SRD5A1 function, leading to a lower level of dihydrotestosterone (DHT) production and SRD5A1 protein suppression[1].
More description
|
|
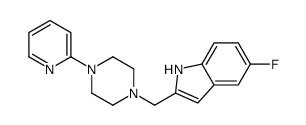
| DC65485 | CP-226269 Featured |
CP 226269 is a potent dopamine D4 receptor agonist that induces calcium flux with EC50 of 32.0 nM. CP 226269 can be used in the research of schizophrenia and other related diseases.
More description
|
|
| DC65486 | ZW290 Featured |
ZW290 is a compound to activate brown adipose tissue (BAT) thermogenic function. ZW290 increases the expression of uncoupling Protein 1 (UCP1) protein and inhibits ATP synthesis in BAT.
More description
|
|
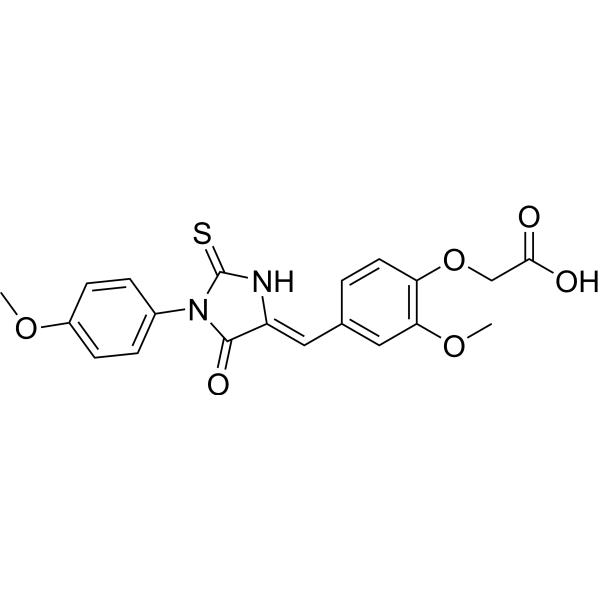
| DC31324 | TRC102 Featured |
Methoxyamine is an orally bioavailable small molecule inhibitor with potential adjuvant activity. Methoxyamine covalently binds to apurinic/apyrimidinic (AP) DNA damage sites and inhibits base excision repair (BER), which may result in an increase in DNA strand breaks and apoptosis. This agent may potentiate the anti-tumor activity of alkylating agents. Check for active clinical trials or closed clinical trials using this agent. (NCI Thesaurus).
More description
|
|
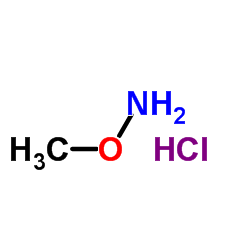
| DC65484 | WAY-639889 Featured |
WAY-639889 is a bioactive compound relative to neuropeptide Y-5.
More description
|
|
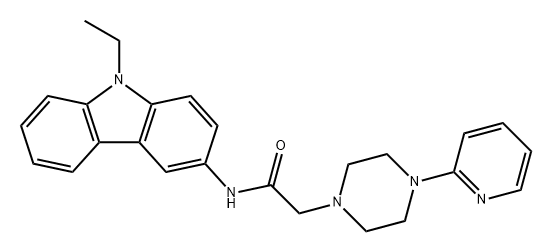
| DC32883 | Levobunolol Hydrochloride Featured |
Levobunolol Hydrochloride is a non-cardioselective adrenergic beta-receptor antagonist with anti-glaucoma activity.
More description
|
|
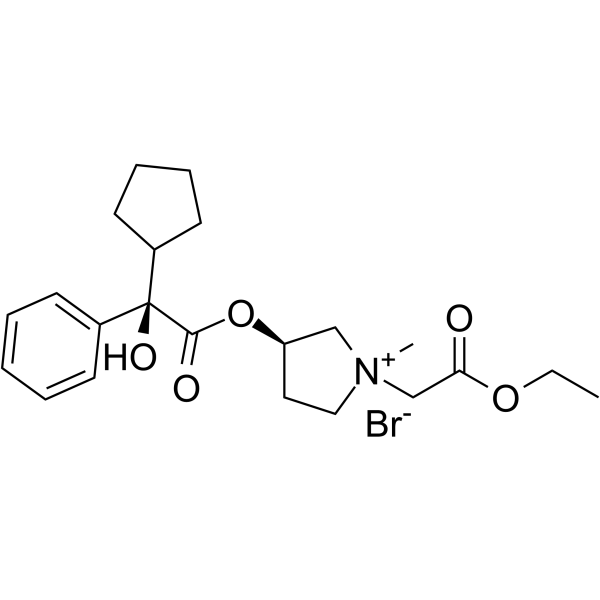
| DC45324 | Sofpironium bromide Featured |
Sofpironium bromide (BBI 4000) is an anticholinergic agent used in the study of primary axillary hyperhidrosis (PAH). Sofpironium bromide reduces sweating by inhibiting M3 muscarinic receptors in eccrine glands at the application site. Sofpironium bromide also has a high afnity for the M1, M2, M4 and M5 subtypes.
More description
|
|
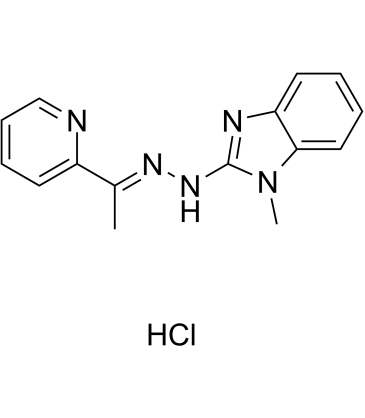
| DC65483 | SI-2 (hydrochloride) Featured |
SI-2 hydrochloride (EPH 116 hydrochloride) is a highly promising SRC-3 SMI: SRC-3 inhibitor (PPI), with IC50 values of 3-20 nM for breast cancer cell death. SI-2 hydrochloride (EPH 116 hydrochloride) has a much improved toxicity and pharmacokinetic profile, with acceptable oral availability[1].
More description
|
|
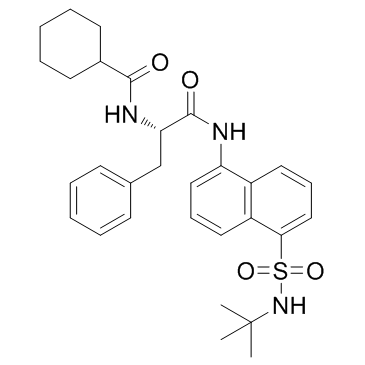
| DC10917 | MF-094 Featured |
MF-094 (MF094) is a potent, highly selective inhibitor of ubiquitin specific protease 30 (USP30) with IC50 of 0.12 uM, demonstrates <30% inhibitory activity for a panel of 22 USPs assays at 10 uM.
More description
|
|
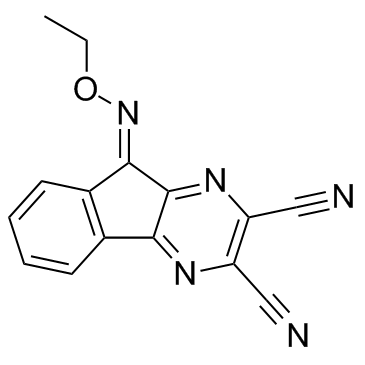
| DC24043 | USP8-IN-22e Featured |
A potent, selective ubiquitin-specific protease USP8 inhibitor with IC50 of 0.28 uM.
More description
|
|
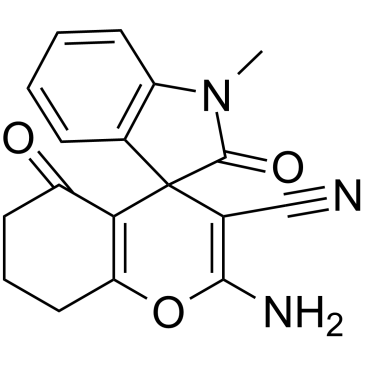
| DC42616 | EMBL Featured |
Novel inhibitor of MuRF1, attenuating skeletal muscle atrophy and dysfunction in cardiac cachexia
More description
|
|
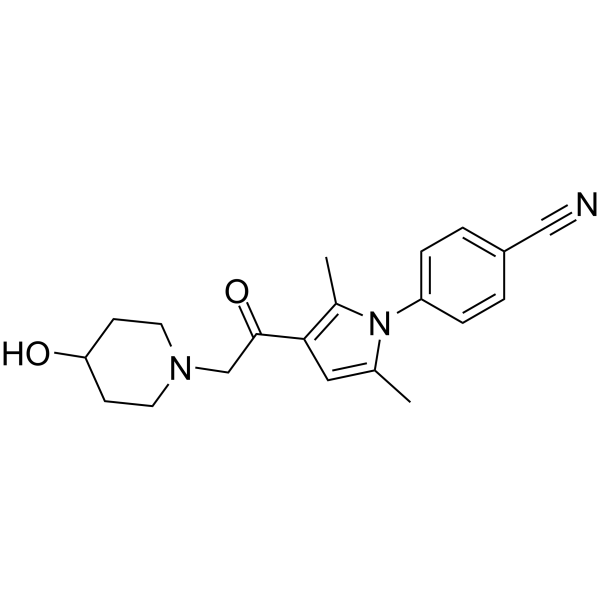
| DC71668 | Iu1-248 Featured |
Iu1-248, a derivative of IU1, is a potent and selective ubiquitin specific peptidase 14 (USP14) inhibitor with an IC50 of 0.83 μM.
More description
|
|
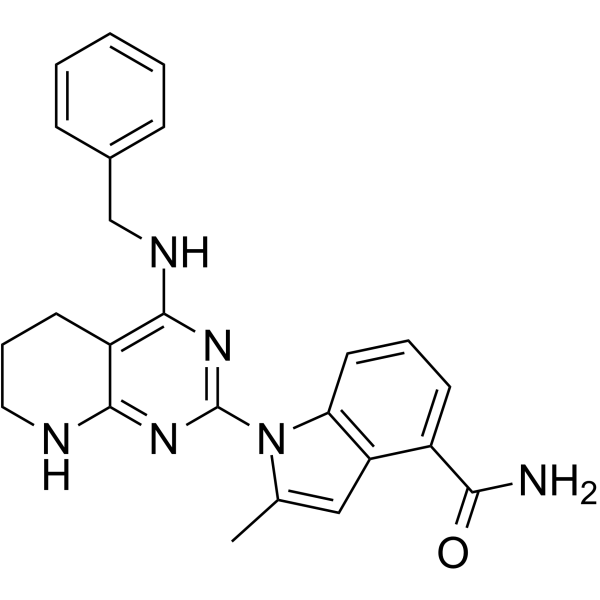
| DC48941 | p97-IN-1 Featured |
p97-IN-1 is a potent p97 inhibitor with an IC50 <30 nM (WO2015109285A1, compound FF07).
More description
|
|
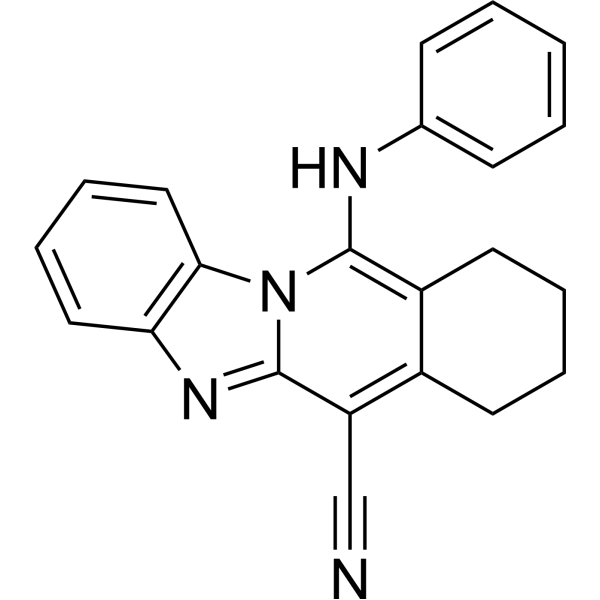
| DC72826 | Usp22i-S02 Featured |
Usp22i-S02(USP22-IN-1, compound S02) is a potent inhibitor of ubiquitin specific peptidase 22 (USP22). It showed anticancer activity by suppressing Foxp3 expression in T-regulatory cells.
More description
|
|
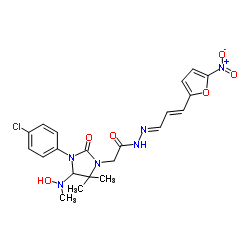
| DC65481 | Eeyarestatin I Featured |
Eeyarestatin I is a potent inhibitor of endoplasmic reticulum-associated protein degradation (ERAD). Eeyarestatin I also inhibits Sec61 translocon.
More description
|
|
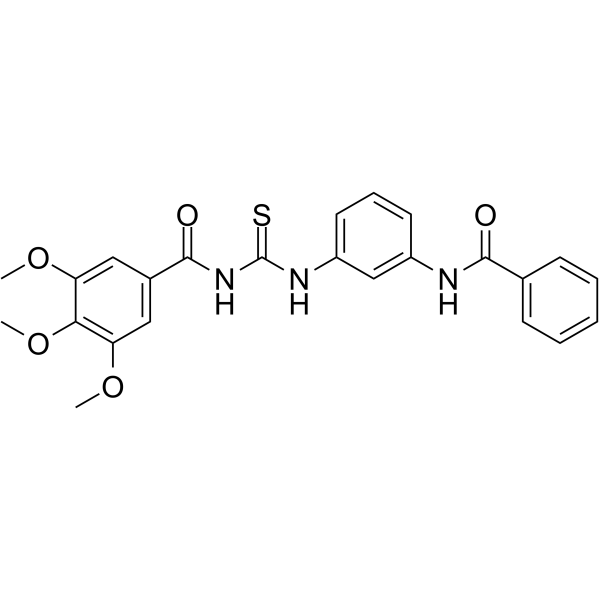
| DC43413 | MRT-10 Featured |
MRT-10 is a seven-transmembrane receptor smoothened (Smo) antagonist with an IC50 of 0.65 μM in the micromolar range in various Hedgehog (Hh) assays. MRT-10 binds to the Smo receptor at the level of the Bodipycyclopamine binding site. MRT-10 can be used for the research of cancer[1][2].
More description
|
|
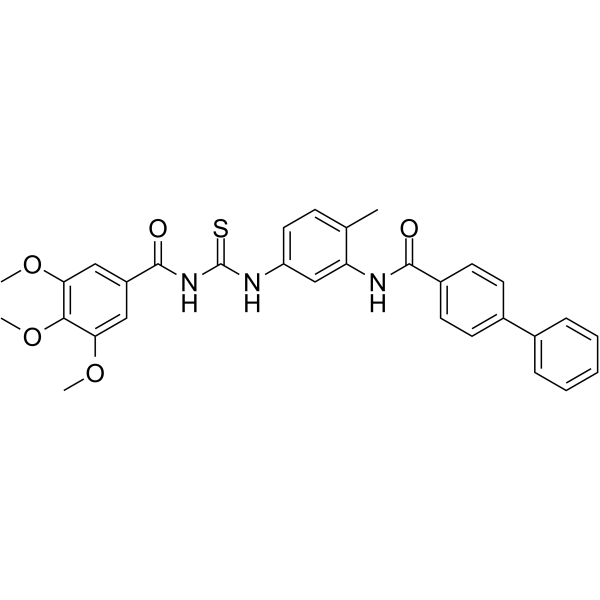
| DC47944 | MRT-81 Featured |
MRT-81 is a potent antagonist of human and rodent smoothened Smo receptors, with an IC50 value of 41 nM in the Shh-light2 cells. MRT-81 has potent hedgehog inhibiting activity. MRT-81 can be used for the research of cancer.
More description
|
|
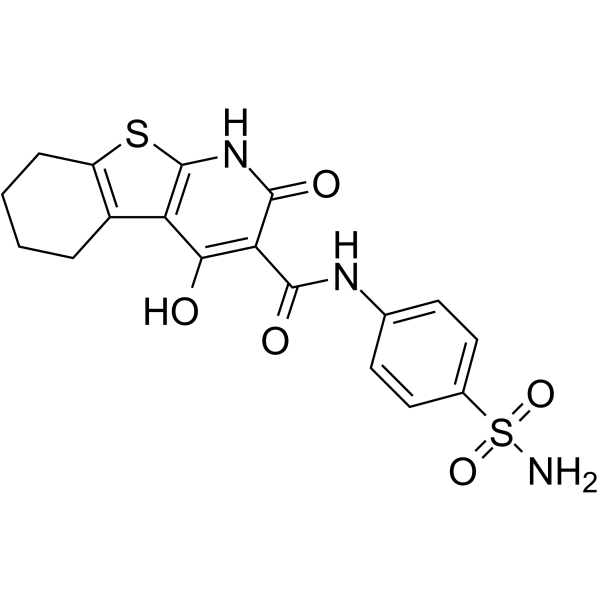
| DC47678 | M435-1279 Featured |
M435-1279 is a UBE2T inhibitor. M435-1279 acts inhibit the Wnt/β-catenin signaling pathway hyperactivation through blocking UBE2T-mediated degradation of RACK1.
More description
|
|
| DC21668 | ALLO-2 Featured |
ALLO-2 is a potent small molecule Smoothened (Smo) antagonist that inhibits Smo agonist Hh-Ag1.5-induced luciferase expression in TM3-Gli-Luc cells with IC50 of 6 nM.
More description
|
|
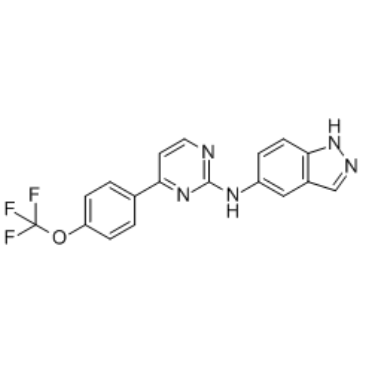
| DC65480 | TPH104m Featured |

|
|
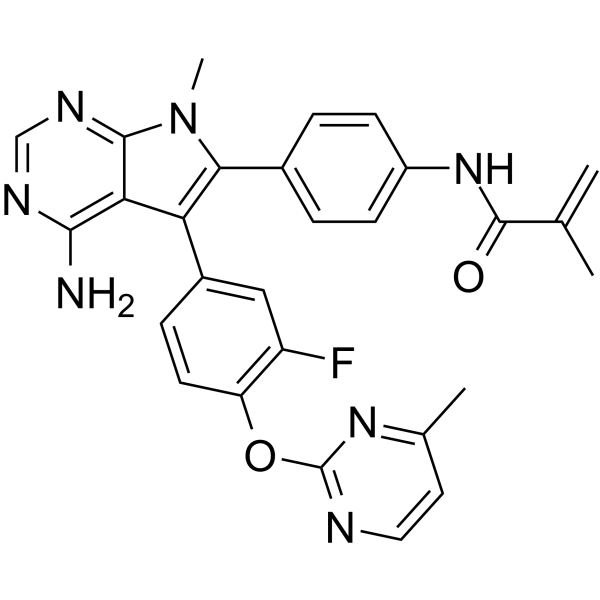
| DC72885 | Lirafugratinib Featured |
Lirafugratinib (RLY-4008) is an orally active and selective inhibitor of FGFR2.
More description
|
|
| DC65477 | Laprituximab emtansine Featured |
Laprituximab emtansine (IMGN-289) is an immunotoxin targeting HER1. Laprituximab emtansine is an EGFR antibody-drug conjugate (ADC) consisting of the J2898A antibody, DM1 (anti-microtubule agent) and the SMCC thioether linker. Laprituximab emtansine can be used for cancer research[1][2][3].
More description
|

|
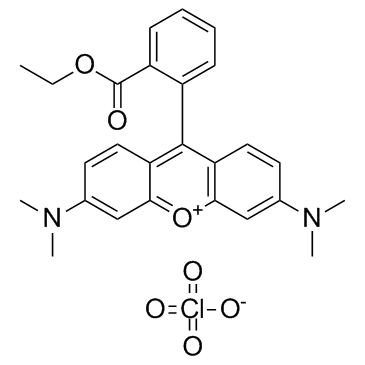
| DC65474 | TMRE Featured |
TMRE is a mitochondria specific dye (λex=550 nm, λem=575 nm).
More description
|
|
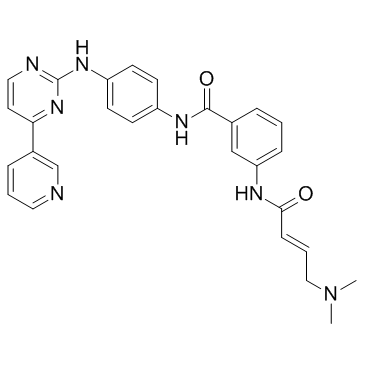
| DC36480 | JNK-IN-7 Featured |
JNK-IN-7 is a relatively selective JNKs inhibitor(IC50= 1.54/1.99/0.75 for JNK1/2/3); also bound to IRAK1, PIK3C3, PIP5K3 and PIP4K2C.
More description
|
|