 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
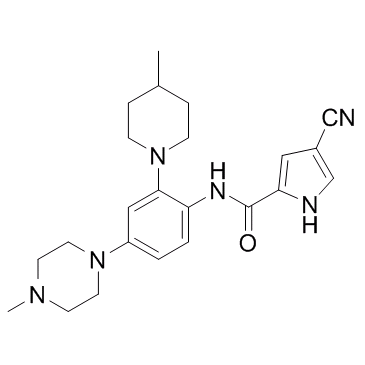
| DC22809 | c-FMS-IN-8 Featured |
c-FMS-IN-8 is a potent, selective c-FMS kinase inhibitor with IC50 of 0.8 nM, shows activity in collagen-induced model of arthritis in mice..
More description
|
|
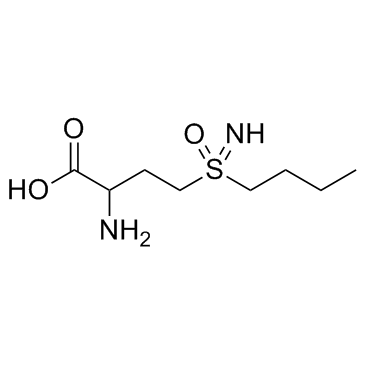
| DC12252 | D,L-Buthionine-(S,R)-sulfoximine Featured |
D,L-Buthionine-(S,R)-sulfoximine is a potent inhibitor of glutamylcysteine synthetase biosynthesis.
More description
|
|
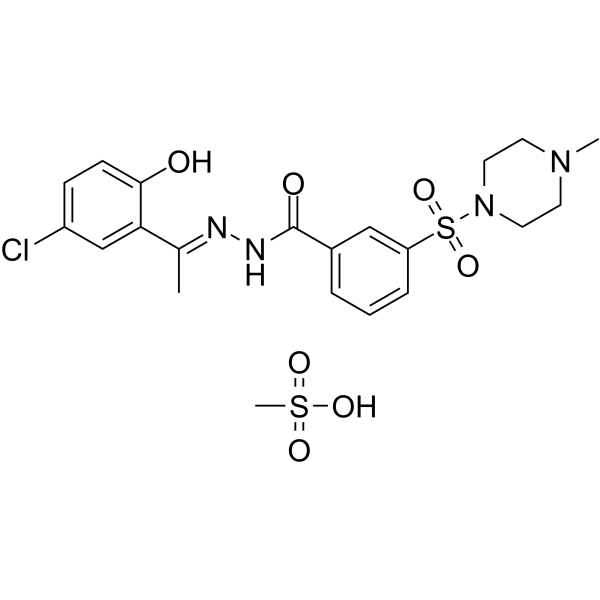
| DC47621 | Seclidemstat mesylate Featured |
Seclidemstat (SP-2577) mesylate is a potent noncompetitive and reversible KDM1A (LSD1) inhibitor (Ki=31 nM, IC50=13 nM). Seclidemstat mesylate promotes antitumor immunity in switch/sucrose nonfermentable (SWI/SNF) complex mutated ovarian cancer, as well as inhibit virus production, viral DNA replication, and late gene expression. Seclidemstat mesylate can be used for the research of Ewing Sarcoma.
More description
|
|
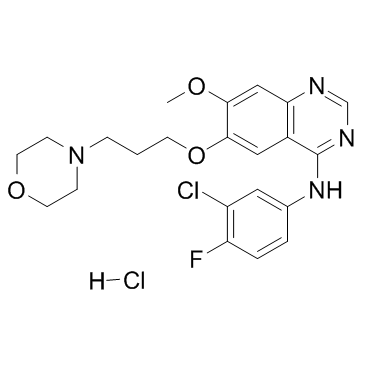
| DC22485 | Gefitinib hydrochloride Featured |
A potent, selective, orally active EGFR inhibitor with IC50 of 23 nM.
More description
|
|
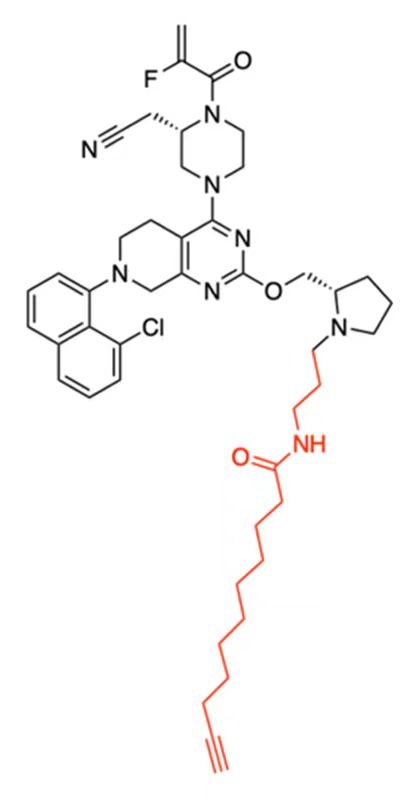
| DC60491 | C11-MRTX Featured |
C11-MRTX is a lipid-conjugated MRTX849 analogue with a 11-carbon tail. C11-MRTX is a nonaggregating potent cellular inhibitor of K-Ras(G12C).
More description
|
|
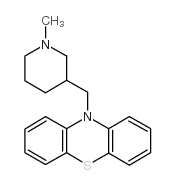
| DC65603 | Mepazine Featured |
Mepazine (Pecazine) is a potent and selective MALT1 inhibitor. Mepazine inhibits GSTMALT1 full length and GSTMALT1 325-760 with IC50s of 0.83 and 0.42 μM, respectively. Mepazine affects viability of ABC-DLBCL cells by enhancing apoptosis.
More description
|
|
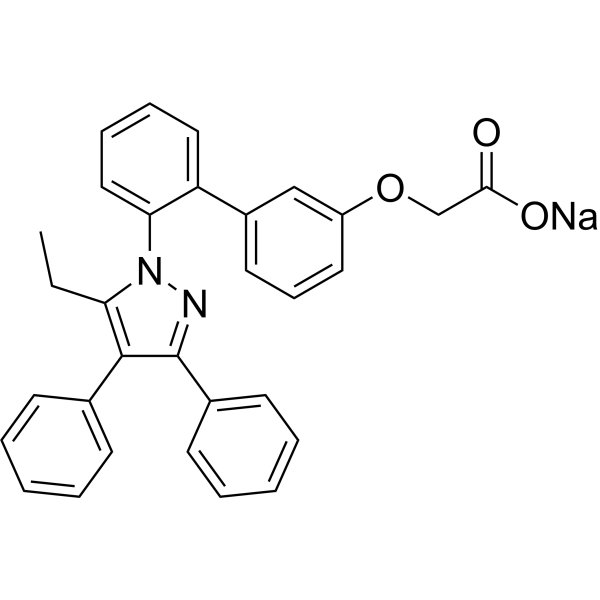
| DC65602 | BMS-309403 sodium Featured |
BMS-309403 sodium is a potent, orally active, and selective adipocyte fatty acid binding protein (also known as FABP4, aP2) inhibitor, with Kis of <2, 250, and 350 nM for FABP4, FABP3, and FABP5, respectively. BMS-309403 sodium interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. BMS-309403 sodium improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells.
More description
|
|
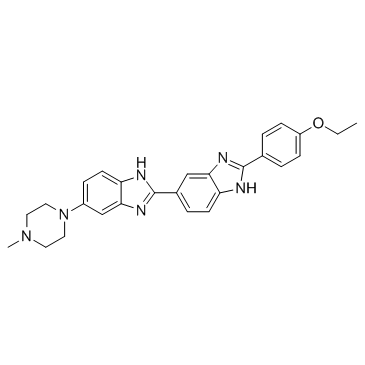
| DC65601 | Hoechst 33342 Featured |
Hoechst 33342 is a DNA minor groove binder used fluorochrome for visualizing cellular DNA.
More description
|
|
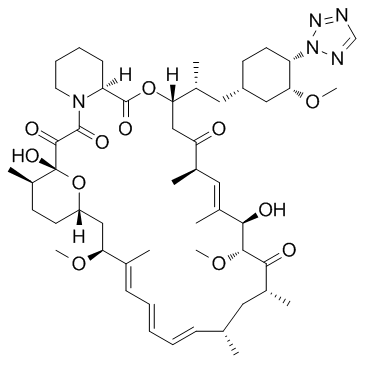
| DC65600 | 42-(2-Tetrazolyl)rapamycin Featured |
42-(2-Tetrazolyl)rapamycin is a prodrug compound of a rapamycin analog extracted from patent US 20080171763 A1, Example 1. Rapamycin is a specific mTOR inhibitor.
More description
|
|
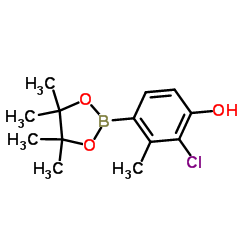
| DC65598 | 2-Chloro-3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Featured |
|
|
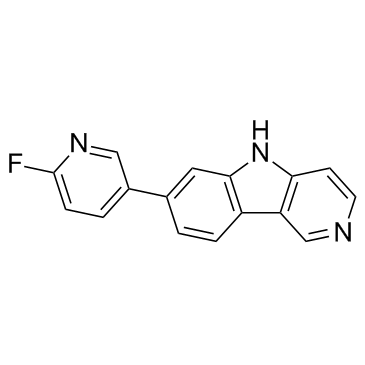
| DC65597 | T807 Featured |
T807 a novel tau positron emission tomography (PET) tracer.
More description
|
|
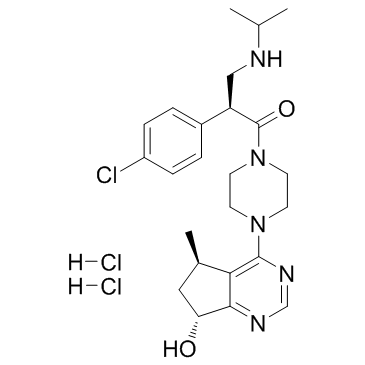
| DC65596 | GDC-0068 (dihydrochloride) Featured |
Ipatasertib dihydrochloride (GDC-0068 dihydrochloride) is a highly selective pan-Akt inhibitor targeting Akt1/2/3 with IC50 of 5/18/8 nM, 620-fold selectivity over PKA.
More description
|
|
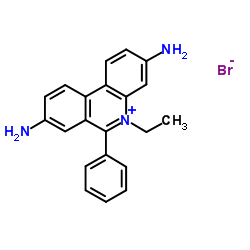
| DC65595 | Ethidium bromide Featured |
Ethidium bromide is an intercalating agent commonly used as a fluorescent tag (nucleic acid stain) in molecular biology laboratories for techniques such as agarose gel electrophoresis.
More description
|
|
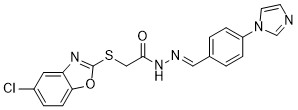
| DC65589 | AKT-IN-18 Featured |
AKT-IN-18, an inhibitor of Akt, inhibits Akt.
More description
|
|
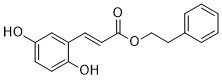
| DC65588 | 5-LOX-IN-2 Featured |
5-LOX-IN-2 is an inhibitor of 5-lipoxygenase (5-LOX).
More description
|
|
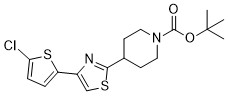
| DC65587 | ARC7 Featured |
ARC7 can act as a probe for secondary metabolism in S. coelicolor.
More description
|
|
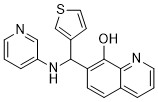
| DC65586 | B-Raf IN 15 Featured |
B-Raf IN 15 (Compound 7) is a BRAF inhibitor.
More description
|
|
| DC65585 | Sotuletinib dihydrochloride Featured |
Sotuletinib, also known as BLZ945, is a potent and selective CSF-1R kinase inhibitor. BLZ945 showed effects of CSF1R inhibition on other tumor-infiltrating immune cells. BLZ945 attenuates the turnover rate of TAMs while increasing the number of CD8+ T cells that infiltrate cervical and breast carcinomas. BLZ945 decreases the growth of malignant cells in the mouse mammary tumor virus-driven polyomavirus middle T antigen (MMTV-PyMT) model of mammary carcinogenesis. BLZ945 prevents tumor progression in the keratin 14-expressing human papillomavirus type 16 (K14-HPV-16) transgenic model of cervical carcinogenesis.
More description
|
.jpg)
|
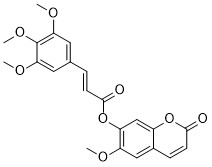
| DC65584 | Antitumor agent-93 Featured |
Antitumor agent-93 (compound 7D) is an anticancer agent.
More description
|
|
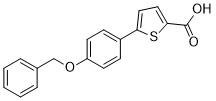
| DC65583 | Nurr1 agonist 2 Featured |
Nurr1 agonist 3 (Compound 7) is a Nurr1 agonist.
More description
|
|
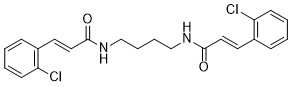
| DC72906 | BCPA Featured |
BCPA is a Pin1 regulator without cytotoxicity. BCPA attenuates the reduction of Pin1 protein to inhibit receptor activator of RANKL-induced osteoclastogenesis. BCPA regulates osteoclast activation, used to osteoporosis research.
More description
|
|
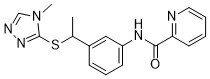
| DC65582 | Cbl-b-IN-5 Featured |
Cbl-b-IN-5 (compound 6) is a Cbl-b inhibitor.
More description
|
|
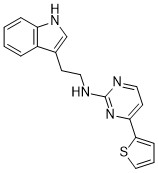
| DC65581 | AP-C2 Featured |
AP-C2 is a potent guanosine 3',5'-cyclic monophosphate (cGMP)-dependent protein kinase II (cGKII) inhibitor.
More description
|
|
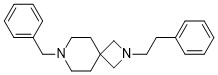
| DC65580 | AD-186 Featured |
AD186 is a potent and selective S1R agonist
More description
|
|
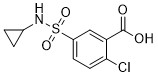
| DC65579 | h-NTPDase8-IN-1 Featured |
h-NTPDase8-IN-1 (compound 2d) is a sulfamoyl-benzamides based and selective inhibitor for h-NTPDases8
More description
|
|
| DC65578 | ROS inducer 1 Featured |
ROS inducer 1 (compound I29) is a fungicide, with EC50 against Xanthomonas axonopodis pv. citri (Xac), Xanthomonas oryzae pv. oryzae (Xoo), and Pseudomonas syringae pv. actinidiae (Psa)
More description
|
|
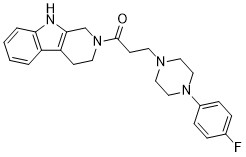
| DC65577 | P-Gb-In-1 Featured |
P-gb-IN-1 (compound Ⅲ-8), a 2,5-disubstituted furan derivative, is a highly effective, broadspectrum P-glycoprotein (P-gp) inhibitor.
More description
|
|
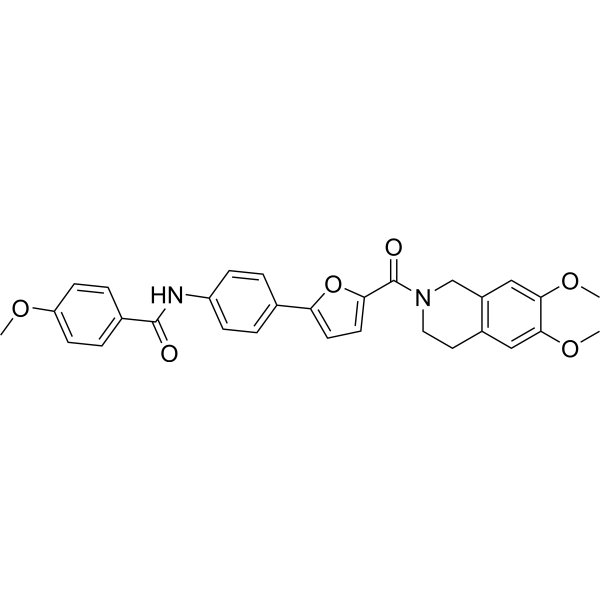
| DC65576 | 17β-HSD10-IN-1 Featured |
17β-HSD10-IN-1 (compound 9) is an orally active inhibitor of 17β-hydroxysteroid dehydrogenase type 10 (17β-HSD10) with blood-brain permeability. 17β-HSD10-IN-1 doesn't result additional effects for mitochondrial off-targets and cytotoxic or neurotoxic effects.
More description
|
|
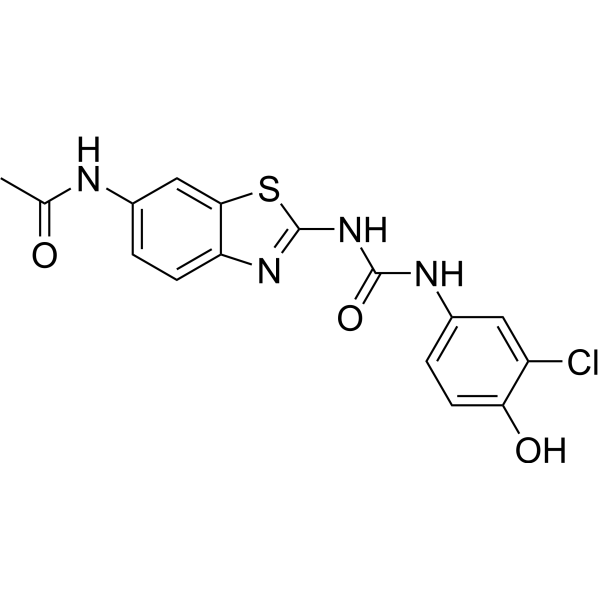
| DC65575 | 17β-HSD10-IN-2 Featured |
17β-HSD10-IN-2 (compound 11) is a benzothiazolylurea-based inhibitor.
More description
|
|
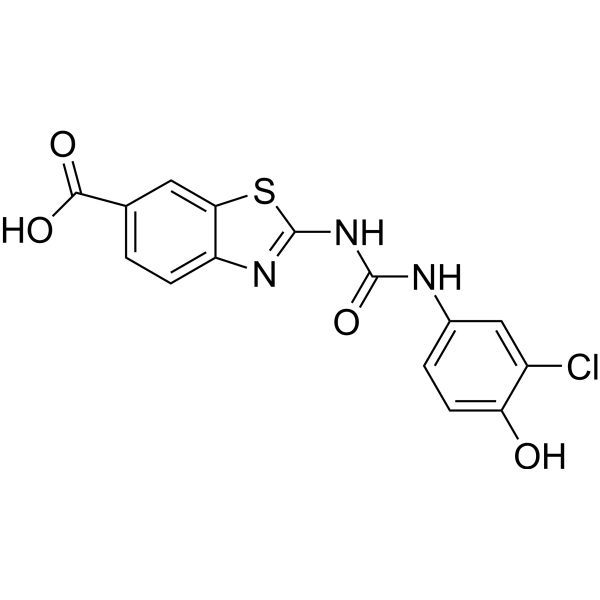
| DC65574 | Anti-NASH agent 1 Featured |
Anti-NASH agent 1 (compound 3d),a derivative of Elafibranor (HY-16737),is a potent agonist of PPAR-α/δ.
More description
|
|