 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
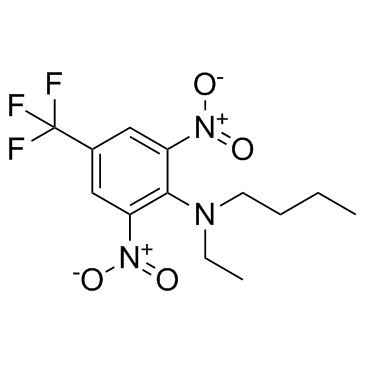
| DC12299 | Benfluralin |
Benfluralin is a kind of herbicide and an agrochemical which can be used as a pre-emergence herbicide used for the control of grass and other weeds in a range of food and non-food crops.
More description
|
|
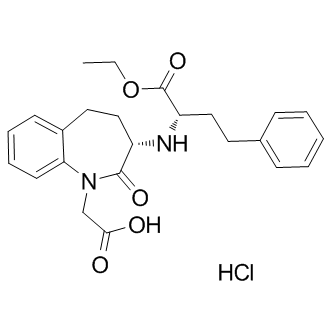
| DC9152 | Benazepril Hydrochloride |
Benazepril hydrochloride, an angiotensin converting enzyme inhibitor, which is a medication used to treat high blood pressure.
More description
|
|
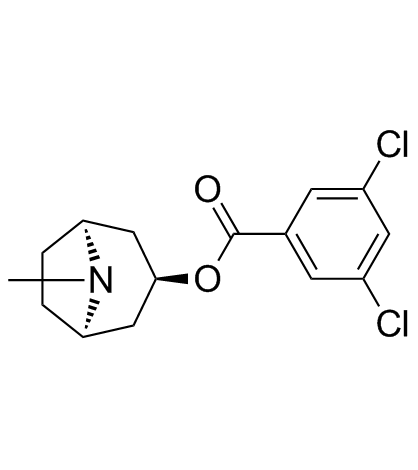
| DC20135 | Bemesetron (MDL 72222) |
Bemesetron (MDL 72222) is a selective 5-HT3 receptor antagonist with an IC50 of 0.33 nM. Neuroprotective effect.
More description
|
|
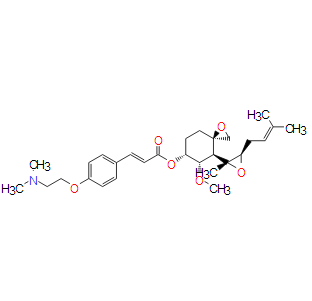
| DC8160 | ZGN-440(Beloranib) |
Beloranib is being studied as a first-in-class obesity therapy that demonstrates a unique mechanism of action through methionine aminopeptidase 2 (MetAP2 ) inhibition.
More description
|
|
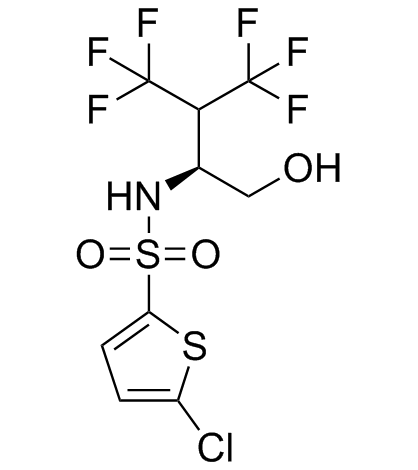
| DC20030 | Begacestat (GSI-953) |
Begacestat (GSI-953) is a selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase (IC50Aβ40=15 nM) for the treatment of Alzheimer's disease.
More description
|
|
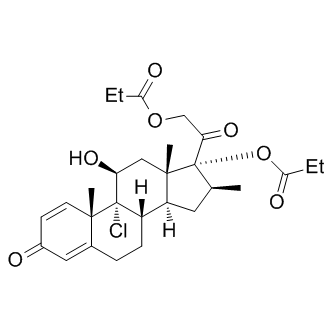
| DC9172 | Beclomethasone Dipropionate |
Beclometasone dipropionate is a potent glucocorticoid agonist; it is a prodrug of the free form, beclometasone.
More description
|
|
| DC20668 | Becampanel |
Becampanel (AMP 397) potent, competitive antagonist of the AMPA receptor (AMPAR) with IC50 of 11 nM.
More description
|

|
| DC20319 | Beauvericin |
Beauvericin is a cyclic hexadepsipeptide with antibiotic and insecticidal effects, shows activity against Gram-positive bacteria and mycobacteria, and is also capable of inducing programmed cell death in mammals..
More description
|
|
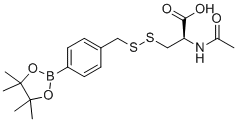
| DC12596 | BDP-NAC |
BDP-NAC is a novel persulfide donor that shows selectivity towards H2 O2 over other potential oxidative or nucleophilic triggers, resulting in the sustained release of the persulfide of N-acetyl cysteine (NAC).
More description
|
|
| DC23980 | BCX-1470 |
BCX-1470 (BCX1470) is a serine protease inhibitor that inhibits the esterolytic activity of factor D (IC50=96 nM) and C1s (IC50=1.6 nM).
More description
|
|
| DC20761 | BCM-599 |
BCM-599 is an efficient HBV capsid assembly inhibitor with in vitro IC50 of 13 uM, shows synergistic inhibitory effects on decreasing viral concentration combined with lamivudine in vivo..
More description
|
|
| DC20760 | BCL6-IN-8 |
BCL6-IN-8 (BCL6-i) is a potent, irreversible and cell-active BCL6 inhibitor with inact/KI value of 1.9 × 104 M-1 s-1, targets Cys53 located at the protein-protein interaction interface.
More description
|
|
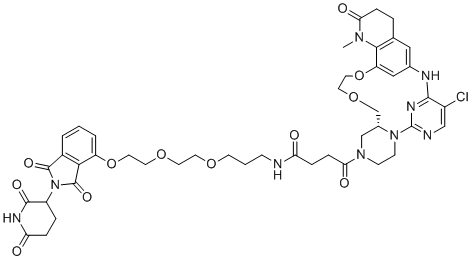
| DC12447 | BCL6 PROTAC 15 |
BCL6 PROTAC 15 is a novel B-cell lymphoma 6 (BCL6) PROTAC, significantly degrade BCL6 in a number of DLBCL cell lines, but neither BCL6 inhibition nor degradation selectively induces marked phenotypic response.
More description
|
|
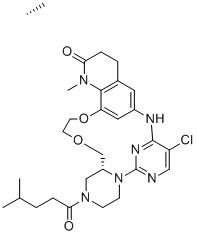
| DC12446 | BCL6 inhibitor 14 |
BCL6 inhibitor 14 is a potent BCL-6 inhibitor with FRET IC50 of 63 nM..
More description
|
|
| DC20758 | BCI-137 |
BCI-137 is a chemical compound able to compete with Argonaute 2 miRNAs binding, binds to Ago2 with Kd of 126 uM.
More description
|
|
| DC20757 | BC-23 |
BC-23 (NSC 45382) is a novel inhibitor of β-catenin/Tcf4 interaction with IC50 of 1.7 uM.
More description
|
|
| DC21401 | BC-21 |
BC-21 (NSC 109268) is a novel small molecule inhibitor that effectively inhibits the β-catenin/Tcf4 interaction with Ki of 6.55 uM.
More description
|
|
| DC20755 | BC-1485 |
BC-1485 is a first-in-class, small molecule inhibitor of FIEL1 (Fibrosis-inducing E3 ligase 1) that exhibits potent activity toward disrupting FIEL1-directed PIAS4 ubiquitination.
More description
|
|
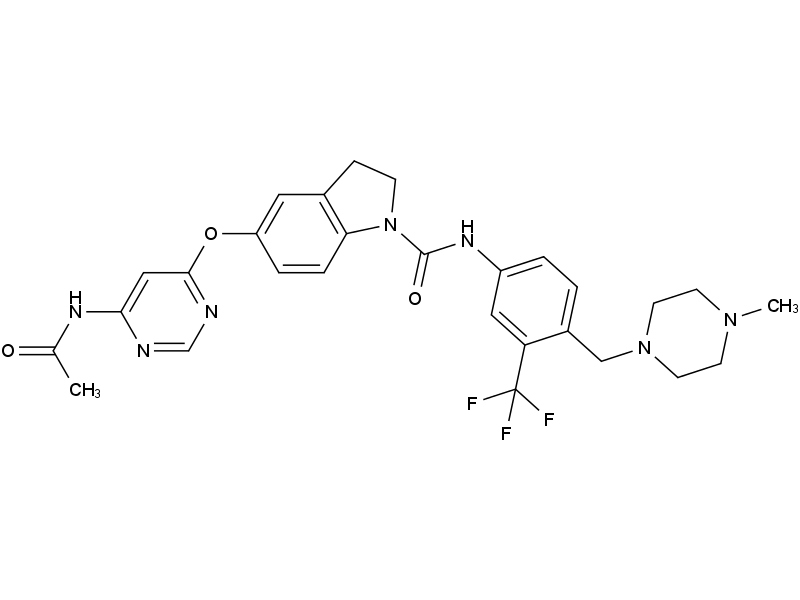
| DC10058 | BBT594 |
BBT594 (NVP-BBT594), is potent and selective inhibitor of RET and JAK2.
More description
|
|
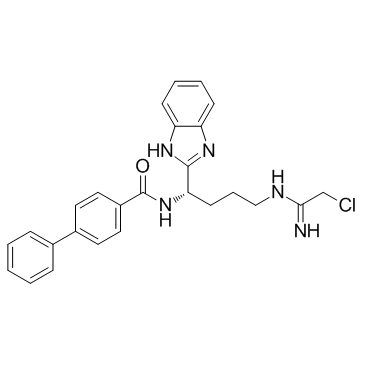
| DC12280 | BB-Cl-Amidine |
BB-Cl-Amidine is a peptidylarginine deminase (PAD) inhibitor.
More description
|
|
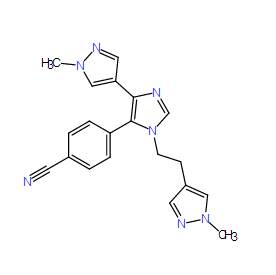
| DC7854 | BAZ2-ICR |
BAZ2-ICR is a selective BAZ2 bromodomain inhibitor (Kd values are 109 and 170 nM for BAZ2A and BAZ2B respectively).
More description
|
|
| DC11915 | BAY-958 |
BAY-958 (BAY958, LDC526) is a potent, selective PTEFb/CDK9 inhibitor with IC50 of 5 nM against CDK9/CyclinT1.
More description
|

|
| DC22017 | BAY-826 |
BAY-826 (BAY826) is a novel selective, highly potent, orally available TIE-2 inhibitor (dissociation constant=1.6 nM).
More description
|
|
| DC22016 | BAY-7598 |
BAY-7598 (BAY7598) is a potent, selective MMP12 inhibitor..
More description
|
|
| DC20749 | BAY-707 |
BAY-707 is a highly potent, selective and substrate-competitive inhibitor of MTH1 (NUDT1) with IC50 of 2.3 nM.
More description
|
|
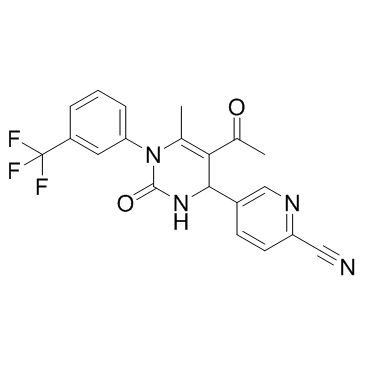
| DC12088 | BAY-678 racemate |
BAY-678 racemate is a chemical probe candidate for Human Neutrophil Elastase (HNE).
More description
|
|
| DC11007 | BAY-524 |
BAY-524 (BAY524) is a potent, selective, ATP-competitive inhibitor of Bub1 kinase with IC50 of 450 nM.
More description
|

|
| DC22015 | BAY-386 |
BAY-386 (BAY386) is a potent, specific and reversible PAR-1 antagonist with IC50 of 10 nM (HEK cell) and binding IC50 of 56 nM, without affinity for PAR-4 (IC50>10 uM).
More description
|
|
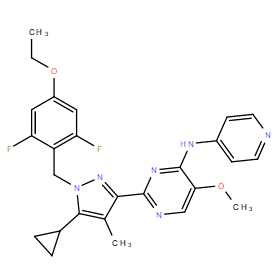
| DC11006 | BAY-320 |
BAY-320 (BAY320) is a potent, selective, ATP-competitive inhibitor of Bub1 kinase with IC50 of 680 nM.
More description
|
|
| DC23816 | BAY-293 racemate |
BAY-293 racemate (BAY293 racemate) is a potent SOS1 inhibitor that disrupts the KRAS-SOS1 interaction with IC50 of 50 nM, the R-enantiomer (BAY-293) is the active form with IC50 of 21 nM, while the BAY-293 is less active (IC50=2,340 nM)..
More description
|
|