 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
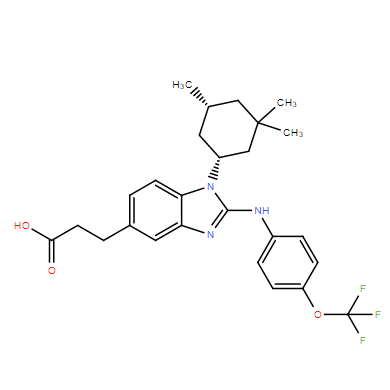
| DC11719 | BAY-1436032 |
BAY-1436032 (BAY1436032) is a potent, selective, orally available inhibitor of pan-mutant IDH1 with IC50 of 15 nM for both IDH1 R132H and R132C, respectively.
More description
|
|
| DC11859 | BAY85-8501 |
BAY 85-8501 (BAY85-8501) is a potent, selective human neutrophil elastase (HNE) inhibitor with IC50 of 65 pM.
More description
|

|
| DC23438 | BAY 60-6583 |
BAY 60-6583 (BAY606583) is a potent, selective adenosine A2B receptor agonist with EC50 of 2.83 nM (murine A2BR).
More description
|

|
| DC20748 | BAY 60-2770 |
BAY 60-2770 is an NO-independent activator of sGC (soluble guanylyl cyclase) with EC50 of 5.4 nM.
More description
|
|
| DC20747 | BAY 41-8543 |
BAY 41-8543 is an NO-independent sGC (soluble guanylyl cyclase) stimulator, inhibits collagen-mediated aggregation in washed human platelets (IC50=90 nM).
More description
|
|
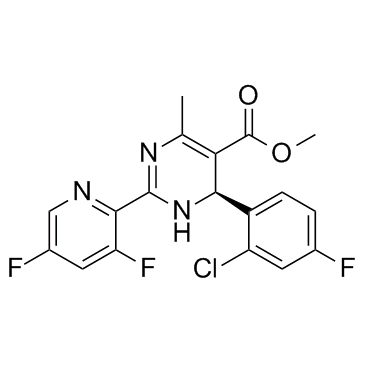
| DC10291 | Bay 41-4109 less active enantiomer |
Bay 41-4109 less active enantiomer shows less activity than Bay 41-4109. BAY 41-4109 is a potent inhibitor of human hepatitis B virus (HBV) with an IC50 of 53 nM.
More description
|
|
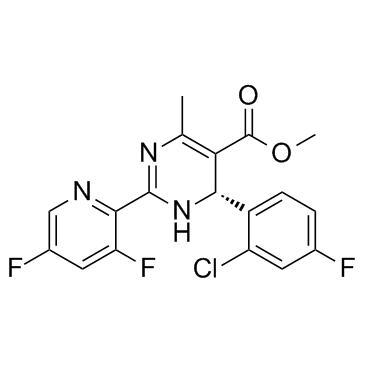
| DC10294 | Bay 41-4109 |
BAY 41-4109 is a potent inhibitor of human hepatitis B virus (HBV) with an IC50 of 53 nM.
More description
|
|
| DC23386 | BAY 1238097 |
BAY 1238097 is a novel BET bromodomain inhibitor with IC50 of 50 nM, inhibits the interaction between BRD4, BRD3 or BRD2 and H4 with IC50 values of 63 nM, 609 nM and 2430 nM in the NanoBRET assay.
More description
|
|
| DC10376 | BAY-1143572 |
BAY 1143572 is a highly selective, potent and orally available inhibitor of PTEFb/CDK9; inhibits the proliferation of AML cell lines with a median IC50 of 385 nM.
More description
|

|
| DC20750 | BAY 1143269 |
BAY 1143269 is a potent, selective, orally bioavailable inhibitor of MNK1 with enzyme IC50 of 40 nM at 2 mM ATP.
More description
|
|
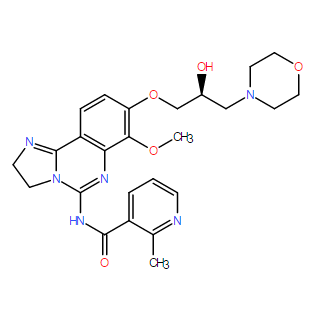
| DC9939 | BAY 1082439 |
BAY 1082439 is a highly selective and balanced PI3Kα/β inhibitor demonstrated potent activity in tumors with activated PI3Kα and loss-of-function of PTEN.
More description
|
|
| DC23719 | BAY 1024767 |
BAY 1024767 (BAY1024767) is a higly potent antagonist of androgen receptor (AR) wild-type (IC50=46 nM) and mutant forms located in the AR ligand-binding domain (LBD).
More description
|
|
| DC20317 | Barbadin |
Barbadin is a small molecule that selectively inhibits the β-arrestin/β2-adaptin interaction (IC50=19.1/15.6 uM for β-arrestin1/2) without effect on the formation of receptor/β-arrestin complexes.
More description
|
|
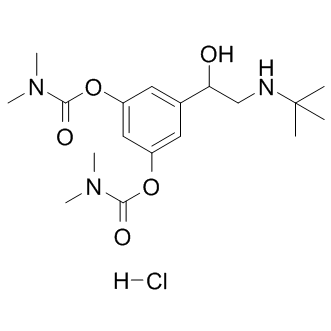
| DC9117 | Bambuterol HCl |
Bambuterol Hcl is a long acting beta-adrenoceptor agonist (LABA) used in the treatment of asthma; it also is a prodrug of terbutaline.
More description
|
|
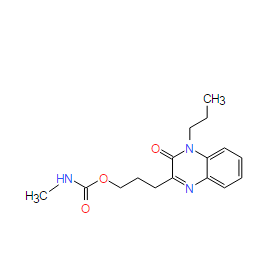
| DC9869 | Bamaquimast |
Bamaquimast is an inhibitor of proton pump extracted from patent US2005165041, example 138.
More description
|
|
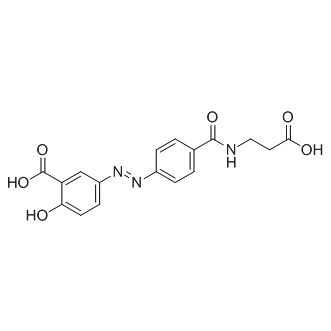
| DC9562 | Balsalazide |
Balsalazide is an anti-inflammatory compound used in the treatment of Inflammatory Bowel Disease.
More description
|
|
| DCAPI1046 | Balofloxacin |
Balofloxacin
More description
|
|
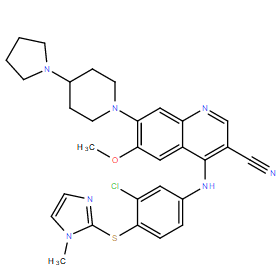
| DC10994 | Balamapimod |
Balamapimod (MKI-833) is an orally active, reversible Ras/Raf/MEK inhibitor developed for antineoplastic potential..
More description
|
|
| DC20740 | BAL30072 |
BAL30072 is a monocyclic beta-lactam antibiotic that shows potent activity against MDR Pseudomonas aeruginosa and Acinetobacter sp. Isolates with MIC90 of 8 and 4 ug/ml.
More description
|
|
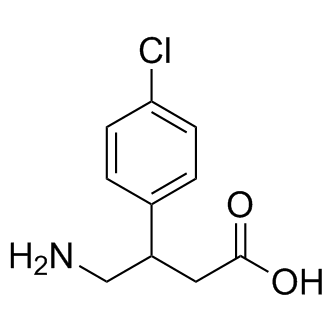
| DC8971 | Baclofen |
Baclofen is a gamma-amino-butyric acid (GABA) derivative used as a skeletal muscle relaxant.
More description
|
|
| DCAPI1066 | Bacitracin zinc |
Bacitracin zinc
More description
|
|
| DC22011 | BAA473 |
BAA473 (BAA-473) is a bile acid analogue that functions as the first small molecule activator of the Pyrin inflammasome, induces secretion of IL18 in LPS-primed PBMCs.
More description
|
|
| DC22010 | B591 |
B591 (B-591) is a potent, specific pan-PI3K inhibitor with IC50 of 1.3/0.36/1.1/1.58 uM for PI3Kα/β/γ/δ, respectively.
More description
|
|
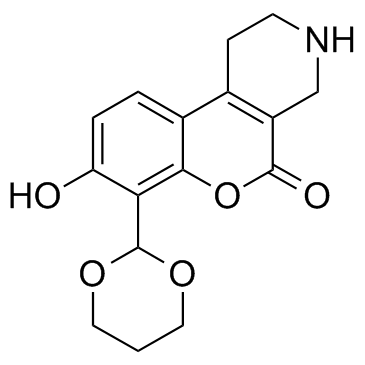
| DC12114 | B I09 |
B I09 is an IRE-1 RNase inhibitor, with an IC50 of 1230 nM.
More description
|
|
| DC23756 | Aβ polymerization stimulator O4 |
Aβ polymerization stimulator O4 is an orcein-related small molecule can drive polymerization of amyloid-β (Aβ).
More description
|
|
| DCAPI1341 | Aztreonam (Azactam, Cayston) |
Aztreonam (Azactam, Cayston)
More description
|
|
| DC22953 | AZSMO-23 |
AZSMO-23 (AZSMO 23, AZSMO23) is a potent hERG potassium channel (Kv11.1) activator that activates WT hERG pre-pulse and tail current with EC50 of 28.6 and 11.2 μM respectively.
More description
|
|
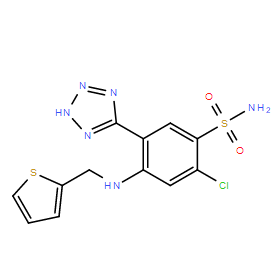
| DC10992 | Azosemide |
Azosemide (Azosemidum) is a potent Na-K-Cl Cotransporter NKCC1 with IC50 of 0.246 and 0.197 uM for hNKCC1A and NKCC1B, being about 4-times more potent than bumetanide.
More description
|
|
| DCAPI1225 | Azomycin (2-Nitroimidazole) |
Azomycin (2-Nitroimidazole)
More description
|
|
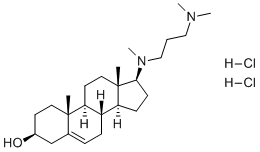
| DC4141 | Azocosterol 2HCL |
Azocosterol is a potential avian reproductive inhibitor.
More description
|
|