| DC70158 |
A293 |
A293 (AVE1231) is a potent and antiarrhythmic compound and selective inhibitor of two-pore-domain potassium channel TASK-1 (KCNK3, hK2P3.1).A293 (AVE1231) is an inhibitor of hKv1.5 currents with predominant action on channels in their open state.A293 (1 uM) prominently depolarized arterial smooth muscle and increased basal tone level and contractile responses to methoxamine of arteries from young rats but had almost no effect in adult rats.Pharmacological inhibition of atrial TASK-1 currents via A293 (AVE1231) exerts antiarrhythmic effects in vivo as well as in silico, resulting in acute cardioversion of paroxysmal trial fibrillation (AF). |

|
| DC70187 |
AM237 |
AM237 is a selective TRPC5 channel activator that potently activated homomeric TRPC5:C5 channels (EC50=15–20 nM in Ca2+i assays).AM237 did not activate TRPC4:C4, TRPC4-C1, TRPC5-C1, TRPC1:C5, and TRPC1:C4 channels, or native TRPC1:C4 channels in A498 cells, but potently inhibited EA‐dependent activation of these channels with IC50 values ranging from 0.9 to 7 nM.AM237 (300 nM) did not activate or inhibit TRPC3, TRPC6, TRPV4, or TRPM2 channels.AM237 potentiated TRPC5:C5 channels activation by sphingosine‐1‐phosphate but suppressed activation evoked by (−)‐englerin A (EA).AM237 concentration‐dependently suppressed further activation of Ca2+ influx mediated by EA with IC50 of 13 nM, potentiated TRPC5:C5 activation by S1P.Pico145 is a competitive antagonist of AM237‐mediated TRPC5:C5 activation. |
|
| DC70201 |
AP-202 |
AP-202 (AP202) is a highly potent and selective α4β2 nAChR antagonist with binding Ki of 18 nM, 57-fold selectivity over α3β4 receptor.AP-202 is agonist activity, also dispalys 40-fold, 10-fold and 90-fold selectivity over α4β4, α3β2, α3β4α5 receptors, does not activate α7 nAChR or block acetylcholine induced changes in membrane potential.AP-202 showed significant activity in blocking nicotine priming-induced as well as cue-induced reinstatement of nicotine seeking in vivo. |
|
| DC70203 |
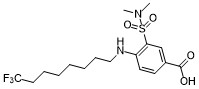
ARN23746
Featured
|
ARN 23746 (ARN-23746) is a potent, selective inhibitor of Na+-K+-Cl- importer NKCC1, shows NKCC1 inhibition 31.8% at 10 uM, and 95.2% at 100 uM in the Cl− influx assay on NKCC1-transfected HEK293 cells.ARN23746 did not show significant NKCC2 inhibition and KCC2 inhibition at 10 uM.ARN23746 selectively blocks NKCC1 in a human cell line and restore the physiological [Cl−]i in murine DS neurons in culture, has excellent solubility and metabolic stability, and displays no issues with off-target activity in vitro.ARN23746 demonstrated in vivo efficacy in rescuing cognitive impairment in a DS mouse model and social deficits and repetitive behaviors in an autism mouse model. |
|
| DC70204 |
ARN23765 |
ARN23765 (ARN 23765) is a highly potent, pharmacological corrector of the mutant CFTR chloride channel with EC50 of 38 pM in cellular assays;
ARN23765 is more than 5000-fold lower compared to presently available corrector drugs.
ARN23765 also showed high efficacy, synergy with other types of correctors, and compatibility with chronic VX-770 potentiator. |
|
| DC70205 |
ARN24092 |
ARN24092 (ARN-4092) is a potent, selective inhibitor of Na+-K+-Cl- importer NKCC1, shows significant dose-dependent inhibition of NKCC1 in the Ca2+ influx assay (51.9% at 100 uM).ARN24092 did not show any significant inhibition of NKCC2 in the Cl-influx assay nor inhibition of KCC2 in the thallium (Tl) influx assay.ARN24092 (i.p, 0.6 mg/kg, daily) restored the short-term working memory of Ts65Dn mice in the T-maze test, completely restored associative memory in Ts65Dn mice in the contextual fear-conditioning (CFC) test, without side effects showed in health of the mice. |
|
| DC70213 |
ASIC1a inhibitor 5b |
ASIC1a inhibitor 5b is a highly selective and potent ASIC1a inhibitor, inhibits proton-evoked ASIC1a currents with an apparent IC50 of 27 nM at pH 6.7.ASIC1a inhibitor 5b can potently and selectively inhibit acid-induced activation of ASIC1a-containing channels, including both ASIC1a homotrimers and ASIC1a-ASIC2 heterotrimers, in a pH-dependent manner.ASIC1a inhibitor 5b binds to the acidic pocket of ASIC1a, exhibits a dramatic leftward shift under pH 6.7 activation of ASIC1a as compared to pH 6.0.ASIC1a inhibitor 5b protected neurons from ASIC1a-dependent cell death and is blood-brain barrier permeable.ASIC1a inhibitor 5b (5 mg/kg) attenuated neurological impairment and alleviated infarct volume in mouse MCAO model of ischemic stroke. |
|
| DC70237 |
Bamocaftor potassium |
Bamocaftor (VX-659) is a next-generation CFTR corrector, restores F508del-CFTR protein function. |
|
| DC70245 |
Benzopyran-G1 |
Benzopyran-G1 is a selective inhibitor of cardiac acetylcholine-activated inwardly rectifying K+ current (IKACh), composed of Kir3.1/Kir3.4 heterotetrameric and Kir3.4 homotetrameric channel subunits. |
|
| DC70247 |
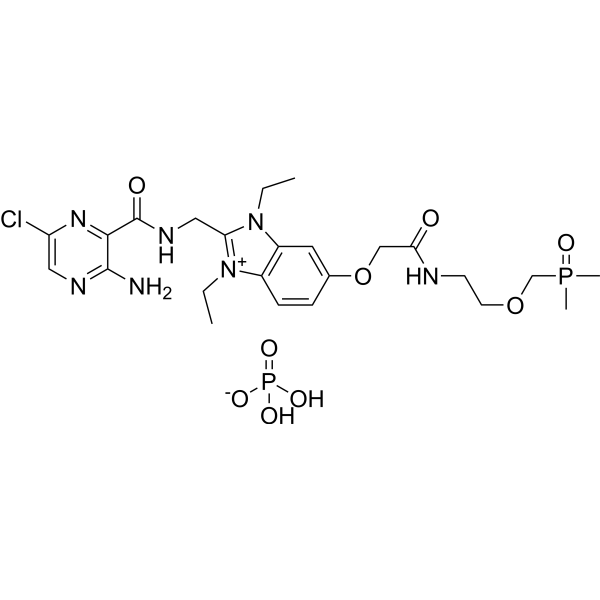
BI 1265162
Featured
|
BI 1265162 is a potent ENaC inhibitor, inhibits Na+ transport with IC50 3 nM and 8 nM in M1 and NCI-H441 cells, respectively.BI 1265162 dose-dependently inhibited Na+ transport and decreased water resorption in cell line models.BI 1265162 reduced liquid absorption in rat lungs and increased MCC in sheep, with no effects on renal function in the animal models.BI 1265162 alone and in combination with CF transmembrane conductance regulator (CFTR) modulators decreased water transport and increased MCC in both normal and CF airway human epithelial models. |
|
| DC70253 |
BI-8668 |
BI-8668 (BI 8668) is a highly potent and selective epithelial sodium channel (ENaC) inhibitor, inhibits Na+ current with IC50 of 17 nM (Ussing chamber assay);
BI-8668 inhibits ENaC-mediated water permeability through cell monolayers of the cell line M-1 (M-1 cells: mouse kidney tubules cells) with 81% inhibition at 3 uM. |
|
| DC70297 |
CDD-1102 |
CDD-1102 (CDD1102) is a potent, selective second bromodomain (BD2) of BRDT and BRD4 inhibitor with IC50 of 7 and 25 nM, >1,000-fold and 300-fold selectivity over BRDT-BD1 and BRD4-BD1. |
|
| DC70312 |
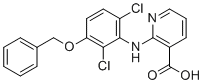
CLC-2-IN-AK42
Featured
|
CLC-2-IN-AK42 (CLC-2 inhibitor AK42, AK42) is a potent, specific small-molecule inhibitor of voltage-gated chloride channel CLC-2 with IC50 of 17 nM (human CLC-2).AK42 displays unprecedented selectivity (>1,000-fold) over CLC-1, the closest CLC-2 homolog, and no off-target engagement a diverse panel of 61 CNS receptors, channels, and transporters expressed in brain tissue.AK-42 binds to an extracellular vestibule above the channel pore. AK-42 is almost three orders-of-magnitude more potent than MCFA, demonstrates high potency (IC50=14 nM for rat CLC-2) in manual patch-clamp experiments on CHO cells transiently transfected with rat CLC-2.AK-42 inhibits hyperpolarization-activated current in hippocampal cells, attenuates steady-state currents and eliminated relaxation currents in recorded neurons.AK-42 is a powerful tool for investigating CLC-2 neurophysiology. |
|
| DC70327 |
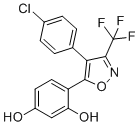
CTIBD
Featured
|
CTIBD is a novel potent activator of the BKCa channel with EC50 of 3.9 uM.CTIBD showed significantly higher potency compared with three other known BKCa activators: NS 1619, NS 11021, and rottlerin.CTIBD induced a reversible potentiation of macroscopic outward currents of the BKCa channel, CTIBD activates both rSlo/rβ1 and rSlo/rβ4 coexpressed channels mainly by decreasing the closing rate of the channel.CTIBD concentration-dependently reduced ACh-induced contractions in isolated rat urinary bladder strips. CTIBD effectively restored frequent voiding contraction and lowered voiding volume without affecting other bladder function parameters in acetic acid-induced overactive bladder (OAB) model (i.p. 20 mg/kg). |
|
| DC70337 |
DA-0218 |
DA-0218 (DA0218) is a novel potent, selective Nav1.7 inhibitor, inhibits sodium currents in Nav1.7-expressing human embryonic kidney 293 cells with IC50 of 0.74 uM.DA-0218 has no effect on sodium currents in Nav1.5-transfected human embryonic kidney 293 cells in patch-clamp experiments.DA-0218 shows analgesic activity predominantly in phase II in formalin-induced inflammatory pain mouse model.DA-0218 produced acute reduction in paclitaxel-induced mechanical allodynia, and inhibited histamine-induced acute itch and lymphoma-induced chronic itch. |
|
| DC70342 |
DCBS152A |
DCBS152A is a potent, functionally selectiver negative modulator of GABAA receptor at the modulatory PQ site in some receptor isoforms. |
|
| DC70367 |
DRB18 |
DRB18 (DRB-18) is a potent, pan-class I glucose transporter (GLUT) inhibitor, reduces glucose uptake in HEK293 cell lines expressd single GLUT1-4 with IC50 of 0.9-8.8 uM.DRB18 reduced cell viability in a dose-dependent manner in cancer cell lines (A549 IC50=3.5 uM, HeLa IC50=1.3 uM), also exhibited IC50 values < 10 μM in all nine melanoma cell lines.DRB18 rapidly and potently inhibited glucose transport and glucose metabolism, inhibited multiple metabolic pathways associated with glucose metabolism in A549 cells.DRB18 caused G1/S phase arrest and increased oxidative stress in A549 cells.DRB18 (10mg/kg) inhibited the growth of A549 tumors xenografted in nude mice. |
|
| DC70379 |
Efonidipine hydrochloride monoethanolate |
Efonidipine (NZ-105) is a potent, dual T-type and L-type calcium channel blocker, exhibits antihypertensive effect through vasodilatation; increases coronary blood flow by blocking L & T-type calcium channels and attenuates myocardial ischaemia, lowers blood pressure in cerebral resistance vessels and prevents hypertension induced brain damage; increases glomerular filtration rate without increasing intra-glomerular pressure and filtration fraction. |
|
| DC70386 |
Encequidar mesylate monohydrate
Featured
|
Encequidar (HM30181) is a potent selective inhibitor of MDR1 (ABCB1, P-gp) with IC50 of 0.63 nM.Encequidar (HM30181) effectively blocked transepithelial transport of paclitaxel in MDCK monolayers.Encequidar (HM30181) does not inhibit MRP1 (ABCC1), MRP2 (ABCC2), and MRP3 (ABCC3), and partially inhibited BCRP (ABCG2) only at very high concentrations.Oral co-administration of paclitaxel and HM30181 showed a tumor-inhibitory strength equal or superior to that of intravenous paclitaxel in the xenograft model in nude mice. |
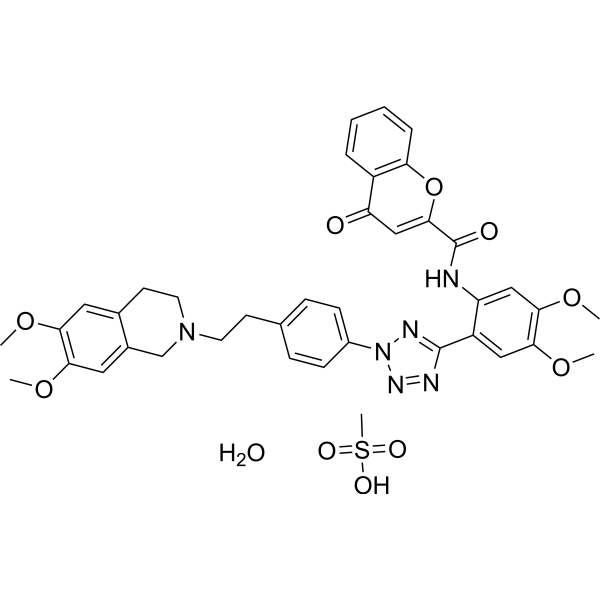
|
| DC70394 |
Etripamil |
Etripamil (MSP-2017) is a novel intranasal non-dihydropyridine calcium channel blocker that has begun phase III clinical trials for the treatment of paroxysmal supraventricular tachycardias. |
|
| DC70426 |
GAT1508
Featured
|
GAT1508 (GAT-1508) is a potent, specific activator for brain-expressed GIRK1/2 channels (EC50=75 nM), specifically activates the brain GIRK1/2 over the cardiac GIRK1/4;
GAT1508 is an allosteric modulator of channel–phosphatidylinositol 4,5-bisphosphate interactions.
GAT1508 effectively extinguished conditioned fear in rodents and lacked cardiac and behavioral side effects, suggesting its potential for use in pharmacotherapy for post-traumatic stress disorder. |
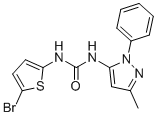
|
| DC70430 |
GDC-0334 |
GDC-0334 (GDC0334) is a highly potent, selective, orally bioavailable TRPA1 antagonist with IC50 of 1.7 nM in cell-based assays.GDC-0334 demonstrated potent TRPA1 inhibition in several species, including human (IC50=1.7 nM), cynomolgus (IC50=3.6 nM), mouse (IC50=2.7 nM), guinea pig (IC50=11.1 nM), and dog (IC50=102 nM).GDC-0334 displays good selectivity against human TRPV1, TRPM8, and TRPC6 (all IC50s>10 uM).GDC-0334 also inhibits calcium flux in human primary cells, HASMCs and HLFs, treated with the TRPA1 agonist AITC.GDC-0334 suppresses AITC-induced edema in vivo in rat (1-10 mg/kg), reduces OVA-induced asthma model in rats and guinea pigs and guinea pig model of cough.GDC-0334 is a potent inhibitor of AITC-induced dermal blood flow (DBF) in vivo in rats and guinea pigs, reduces AITC-induced perfusion and nocifensive behavior in rats and itch and pain scores in humans. |
|
| DC70433 |
Glutor |
Glutor (Glucose uptake inhibitor Glutor) is a novel highly potent glucose uptake inhibitor (IC50=10.8 nM) that selectively targets glucose transporters GLUT-1, -2, and -3.Glutor reduced the uptake of 2-DG with similar potency in different cancer cell lines such as HCT116 (IC50 =10.8 nM), UM-UC-3 (IC50=8.3 nM), UO31 (IC50=3.6 nM), and MIA PaCa-2 (IC50=1.1 nM).Glutor did not interfere with cellular hexokinase activity and potently reduced glycolytic flux in HCT116 cells.Glutor induced upregulation of GLUT-1 and -3 in cancer cells, inhibited glycolysis and efficiently suppresseed the growth of various cancer cell lines.Glutor potently and synergistically inhibited colon cancer cell growth combined with glutaminase inhibitor CB-839. |
|
| DC70451 |
GSK205 derivative 16-8 |
GSK205 derivative 16-8 is a specific small molecule dual-inhibitor of TRPV4 and TRPA1 with IC50 of 0.45 and 0.41 uM, respectively; displays no inhibitory potency toward TRPV1, TRPV2 and TRPV3; effectively attenuates formalin-evoked trigeminal irritant pain in model of acute pancreatitis. |
|
| DC70460 |
GSK3395879
Featured
|
GSK3395879 (GSK-3395879) is a novel potent, selective, orally bioavailable antagonist of TRPV4 with IC50 of 1 nM (hTRPV4); exhibits little or no activity against a broad panel of TRP channels (IC50 > 10µM for TRPA1, TRPV1, TRPM2, TRPM4, TRPM8, TRPC3, TRPC4, TRPC5, TRPC6); demonstrates the ability to inhibit TRPV4-mediated pulmonary edema in an in vivo rat model. |
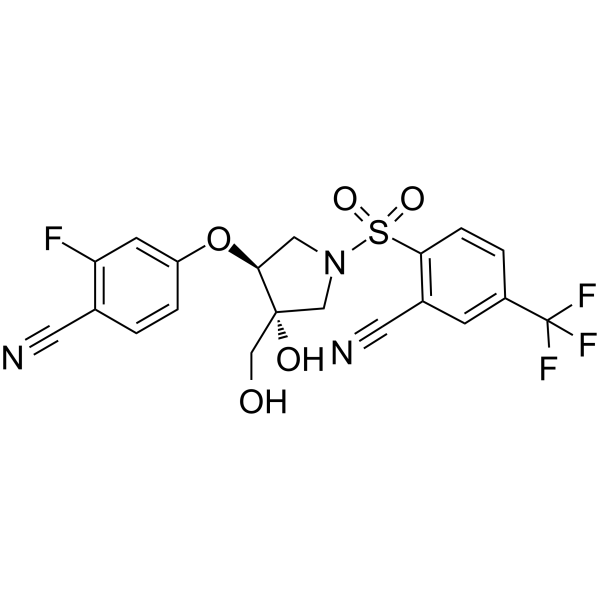
|
| DC70461 |
GSK3527497
Featured
|
GSK3527497 (GSK-3527497) is a potent, selective TRPV4 inhibitor with IC50 of 12 nM (hTRPV4).GSK3527497 is a pre-clinical candidate for treatment of diseases resulting from TRPV4 activation. |
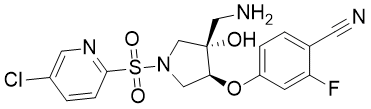
|
| DC70467 |
GSK-5498A |
GSK-5498A is a potent, selective small molecule blocker of Calcium-Release Activated Calcium (CRAC) channel with IC50 of 1 uM; completely inhibits calcium influx through CRAC channels, inhibits mediator release from mast cells, and pro-inflammatory cytokine release from T-cells from multiple human and rat preparations. |
|
| DC70477 |
HCN4 inhibitor EC18 |
HCN4 inhibitor EC18 (EC18) is a HCN4-prefering blocker. |
|
| DC70481 |
HEI3090 |
HEI3090 (HEI-3090) is a small-molecule P2RX7 activator (positive modulator) of purinergic P2RX7 receptor (P2X7R), enhances the P2RX7-mediated intracellular calcium concentration (Emax=250 nM).HEI3090 inhibits tumor growth and combined with αPD-1 immunotherapy ameliorates mice survival.Dendritic cells (DCs) mediate the antitumor activity induced by HEI3090.HEI3090 induces the production of mature IL-18 in the presence of eATP, triggers antitumor responses mediated by IL-18-induced NK and CD4+ T cells.HEI3090 combined with αPD-1 induces antitumor memory immune response. |
|
| DC70500 |
Hv1 inhibitor HIF |
Hv1 inhibitor HIF is a novel inhibitor of the voltage-gated proton channel Hv1 interacts with the Hv1 VSD in the up and down states.HIF rapidly inhibits proton conduction in the up state by blocking the open channel.HIF interacts with a second site that is accessible in the down state and is responsible for key features of HIF-mediated inhibition, such as the slow component of current decay and the slow recovery from inhibition.The voltage-gated proton channel Hv1 plays important roles in numerous biological processes, including pH homeostasis, the immune response, and sperm cell function.Hv1 channel belongs to the large family of proteins containing voltage-sensing domains (VSDs), which also includes Nav, Kv, and Cav channels and voltage-sensitive phosphatases. |
|