| DC70523 |
JNJ-28583113
Featured
|
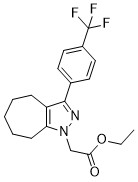
JNJ-28583113 (JNJ28583113) a potent, selective, brain penetrant TRPM2 antagonist with IC50 of 126 nM (hTRPM2).JNJ-28583113 shows similar potency against species chimpanzee and rat TRPM2 with IC50 of 100 and 25 nM, respectively.JNJ-28583113 shows no significant (IC50>10 uM) reactivity towards PARP or PARG and a panel for multiple known kinases GPCRs and ion channels, nine other TRP channels with exception of TRPM5 (IC50<1 uM).Blocking TRPM2 via JNJ-28583113 caused phosphorylation of GSK3α and β subunits in cell assays.JNJ-28583113 also protected cells from oxidative stress induced cell death as well as morphological changes induced by non-cytotoxic concentrations of H2O2。JNJ-28583113 blunted cytokine release in response to pro-inflammatory stimuli in microglia.JNJ-28583113 was brain penetrant but not suitable for systemic dosing as it was rapidly metabolized in vivo, but the in-vitro pharmacology of JNJ-28583113 is the best in class. |
|
| DC70531 |
JW-65 |
JW-65 (JW65) is a selective, potent,CNS-permeable TRPC3 inhibitor improved stability compared to Pyr3.JW-65 shows similar potency and selectivity on TRPC3 channels, but is metabolically much more stable than its precursor, demonstrated by its much longer half-life (>4 h) in mouse, rat, and human liver microsomes when compared to Pyr3.JW-65-treated mice showed substantially decreased susceptibility to PTZ-induced seizures in a dose-dependent manner. |

|
| DC70542 |
KNa1.1 inhibitor 31 |
KNa1.1 inhibitor 31 is a potent, selective, orally available inhibitor of sodium-activated potassium channel KNa1.1 (Slack, Slo2.2) with IC50 of 40 nM (hKNa1.1 WT).KNa1.1 inhibitor 31 reduced seizures and interictal spikes in a mouse model of KCNT1 GoF. |
|
| DC70544 |
KPR-5714 |
KPR-5714 (KPR5714) is a novel potent, selective TRPM8 antagonist with IC50 of 25.3 and 22.4 nM against hTRPM8 and rTRPM8, respectively.KPR-5714 dispalys 400-fold against human TRPA1, TRPV1, and TRPV4, and does not show inhibitory effects for ASIC1a, ASIC3, Nav1.3, Nav1.5, Nav1.6, Nav1.7, and Nav1.8 (IC50 values >10 uM).KPR-5714 (i.p.) inhibited the hyperactivity of mechanosensitive C-fibers of bladder afferents and dose-dependently increased the intercontraction interval shortened by intravesical instillation of acetic acid in anesthetized rats.KPR-5714 (orally administered) dose-dependently increased the mean voided volume and decreased voiding frequency without affecting total voided volume in vivo. |
|
| DC70552 |
Kv2.1-syntaxin inhibitor 15 |
Kv2.1-syntaxin inhibitor 15 (Kv2.1-syntaxin-IN-15, Cpd5) is a small molecule Kv2.1-syntaxin-binding inhibitor (IC50=5.5 uM) with neuroprotective properties.Kv2.1-syntaxin inhibitor 15 competitively binds syntaxin against C1aB-containing Kv2.1 peptides with IC50 of 5.5 uM.Kv2.1-syntaxin inhibitor 15 (10 uM) significantly diminished threo-β-benzyloxyaspartate (TBOA, 75 uM)-induced toxicity neuronal cultures, ameliorated significant cellular damage, by preventing the expression of enhanced Kv2.1-mediated K+ currents.Kv2.1-syntaxin inhibitor 15 is a first-in-class inhibitor of Kv2.1 binding to Syntaxin, likely due to inhibition of the C1a region of Kv2.1 binding to syntaxin.Cpd5 (10 μM) does not affect evoked AMPAR EPSCs in layer 2/3 corticocallosal neurons in the mouse auditory cortex. |
|
| DC70574 |
Lu AA41178
Featured
|
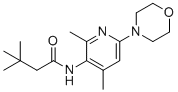
Lu AA41178 is a potent, pan-selective Kv7.2-7.5 opener with EC50 of 1.5, 0.6 20.5, and 37.0 uM for Kv7.2, Kv7.2/7.3, Kv7.4 and Kv7.5, respectively.Lu AA41178 displays no activity at Kv7.1 channels, has no potentiating impact on currents through GABAA ion channels, and a clean off-target profile against common cardiac ion channels.Lu AA41178 significantly increased the seizure thresholds in mice, demonstrating anticonvulsant efficacy in the maximum electroshock seizure threshold test and PTZ seizure threshold test.Lu AA41178 demonstrated antipsychotic-like activity by reducing amphetamine-induced hyperlocomotion in mice as well as lowering conditioned avoidance responses in rats, significantly reduced immobility in mouse model with antidepressant predictivity. |
|
| DC70586 |
MC-100093 |
MC-100093 (MC100093) is a potent, orally bioavailable upregulator of GLT-1 and devoid of antimicrobial properties.MC-100093 enhanced uptake of glutamate into astrocytes in a mouse neuron/astrocyte co-culture model with IC50 of 0.1 uM.MC-100093 restored the expression of NA core GLT-1.MC-100093 (50 mg/kg) attenuated cue-primed relapse to cocaine-seeking without reducing motivation to consume regular chow or sucrose pellets or affecting body weight. |
|
| DC70603 |
ML204 hydrochloride |
ML204 is a potent, selective antagonist of TRPC4 and TRPC5 channels, inhibits TRPC4β-mediated intracellular Ca(2+) rise with IC50 of 0.96 uM, exhibits 19-fold selectivity against TRPC6 channel; blocks TRPC4β currents activated through either μ-opioid receptor stimulation (50 nm DAMGO, IC50=3.55 uM) or intracellular dialysis of GTPγS (IC50=2.85 uM); shows no appreciable block by 10-20 uM for TRPV1, TRPV3, TRPA1, and TRPM8, as well as KCNQ2 and Nav Channels; also inhibits TRPC5 channel currents activated through co-stimulation of Gi/o and Gq/11 signaling by μ-opioid and M3-like muscarinic receptors. |
|
| DC70604 |
ML2-SA1 |
ML2-SA1 is a potent, selective and efficacious activator (agonist) of TRPML2 with EC50 of 1.24 uM and 2.38 uM for human and mouse TRPML2, respectively.ML2-SA1 displays high selectivity for TRPML2 and no significant activity at the related TRPML1 and TRPML3 channels.ML2-SA1 directly stimulates release of the chemokine CCL2 from macrophages, also stimulates macrophage migration, thus mimicking CCL2 function. |
|
| DC70639 |
NaV1.7 inhibitor 51 |
NaV1.7 inhibitor 51 is a highly potent, selective, orally active NaV1.7 inhibitor with IC50 of 10.7 nM in FLIPR membrane potential assay, no significant activity against NaV1.5 (IC50>10 uM).NaV1.7 inhibitor 51 shows good selectivity against other NaV isoforms and minimal inhibition at 10 uM for the resting state across different NaV isoforms.NaV1.7 inhibitor 51 exhibits state-dependent block of human, monkey, and mouse NaV1.7 with a similar potency range.NaV1.7 inhibitor 51 shows significant effects on the CCI-induced neuropathic pain model. |
|
| DC70641 |
NBI-98782 |
NBI-98782 (alpha-dihydrotetrabenazine) is a potent, selective vesicular monoamine transporter (VMAT2, SLC18A2) inhibitor with Ki of 0.97 nM.Acute NBI-98782 decreased mPFC, dSTR, hippocampus, and NAC DA, 5-HT, and NE efflux, while increasing that of DOPAC, HVA, and 5-HIAA.NBI-98782 suppressed clozapine-, olanzapine- and risperidone-induced DA efflux in both mPFC and dSTR, and ACh efflux in mPFC.NBI-98782 attenuated PCP-induced DA, 5-HT, NE and Glu efflux, and AMPH-induced DA and NE efflux, in both mPFC and dSTR, as well as PCP- and AMPH-induced hyperlocomotion. |
|
| DC70658 |
NP10679 |
NP10679 (NP 10679) is a potent, orally active, NMDA receptor subunit 2B (GluN2B)-selective inhibitor of NMDA receptor with IC50 of 23 nM, with no significant effect on GluN2A/C/D (IC50>100 uM).NP10679 demonstrates high pH sensitivity, NP10679 exhibits both a potent IC50 at pH 6.9 of 23 nM with a ratio of IC50 at pH 7.6 to that at pH 6.9 (referred to as pH Boost) of 6.2 fold.NP10679 reduced infarct volume n a dose-dependent manner with an ED50 of 1 mg/kg IP dose and a maximum infarct volume reduction of 52% in MCAo experiments.NP10679 perturbed motor coordination or function in mice. |
|
| DC70685 |
PF-06526290 |
PF-06526290 is a potent, selective Nav1.3 inhibitor with IC50 of 5.1 uM, interact with the Domain 4 voltage sensor domain (D4 VSD and shows no activity for Nav1.7. |
|
| DC70706 |
PRAX-562 |
PRAX-562 (PRAX562) is a novel persistent sodium current (INa) inhibitor, inhibits hNaV1.6 persistent INa induced by ATX-II or SCN8A mutation N1768D with IC50 of 141 and 75 nM, respectively.PRAX-562 displays similar potency for inhibition of persistent INa expressed by other human NaV isoforms (hNaV1.1, hNaV1.2, hNaV1.5) as well as rat, dog, and mouse orthologs (rNaV1.2, dNaV1.2, mNaV1.6, rNaV1.6), with IC50 values ranging 109-180 nM.PRAX-562 exhibits tonic block with lower potency (IC50=8470 nM), demonstrating 60-fold preference for persistent INa, also exhibits preference for persistent INa over peak INa tonic block for hNaV1.1 (173-fold).PRAX-562 reduces intrinsic excitability of hippocampal CA1 pyramidal neurons without compromising action potential (AP) amplitude.PRAX-562 (3 mg/kg, po) produces dose-dependent protection (increase in latency) of mice against MES-induced tonic hindlimb seizures (EC50=90.1 ng/ml), with complete protection at 10 mg/kg. |
|
| DC70712 |
Psalmotoxin 1 |
Psalmotoxin 1 (PCTX1) is a potent and selective acid-sensing ion channel 1a (ASIC1a) blocker with IC50 of 0.9 nM.Psalmotoxin 1 displays no effect at ASIC1b, ASIC2a, ASIC3, heteromeric ASIC channels, ENaC and KV2.1/2.2/4.2/4.3 channels expressed in oocytes at 100 nM.Psalmotoxin 1 exhibits potent analgesic properties against thermal, mechanical, chemical, inflammatory and neuropathic pain in rodents. |
|
| DC70735 |
RGM079 |
RGM079 (RGM-079) is a potent, selective α7 nAChR positive allosteric modulator (PAM) with EC50 of 8.3 uM.RGM079 exhibited a balanced pharmacokinetic profile and antioxidant properties comparable or even higher than well-known natural polyphenols.RGM079 shows neuroprotective properties in Alzheimer's disease (AD)-toxicity related models.RGM079 causes a concentration-dependent neuroprotective effect against the toxicity induced by okadaic acid (OA) in the human neuroblastoma cell line SH-SY5Y.RGM079 is able to restore the cellular viability after exposure to OA and amyloid peptide Aβ1–42, with cell death almost completely prevented at 10 and 30 μM, respectively, in primary cultures of rat cortical neurons;RGM079 shows in vivo analgesic activity in the complete CFA-induced paw inflammation model after intraperitoneal administration. |
|
| DC70736 |
RGM8-51 |
RGM8-51 is a β-lactam derivative, potent, selective TRPM8 antagonist with IC50 of 1.06 and 1.74 uM for rat and human TRPM8, respectively.RGM8-51 does not have any ability to activate TRPM8 channels, but produces a concentration-dependent inhibition of menthol activation.RGM8-51 is selective for cold-activated TRPM8 channels, displays no agonist or antagonist profile in cool-activated TRPA1 channels and has negligible activity as TRPV3 and ASIC3 channel antagonists.RGM8-51 (1 µM) inhibited menthol-activated currents in rTRPM8-expressing HEK293 cells.RGM8-51 exhibits in vivo antinociceptive activity in response to cold, in a mouse model of oxaliplatin-induced peripheral neuropathy.RGM8-51 reduces cold, mechanical and heat hypersensitivity in a rat model of neuropathic pain arising after chronic constriction of the sciatic nerve.RGM8-51 exhibits mechanical hypersensitivity-relieving activity, in a mouse model of NTG-induced hyperesthesia. |
|
| DC70738 |
RJR-2403 hemioxalate |
RJR-2403 (Rivanicline, Metanicotine) is a potent, selective neuronal nicotinic ACh (nAChR) agonist with high affinity to rat brain cortex (Ki=26 nM), does not significantly activate muscle type nAChRs or muscarinic receptors; RJR-2403 is comparable to nicotine in activating rat thalamic synaptosomes with EC50 of 732 nM; significantly improves passive avoidance retention after scopolamine-induced amnesia and enhanced both working and reference memory in rats, also demonstrates greatly reduced cardiovascular effects; exerts its antinociceptive effect via nicotinic rather than either opioid or muscarinic mechanisms. |
|
| DC70774 |
SH045 |
SH045 (SH-045, Larixyl-6-N-methylcarbamate) is a potent, selective TRPC6 antagonist (blocker) with IC50 of 63 nM.SH045 displays 13-fold subtype selectivity over TRPC3 in stably expressing HEK293 cells.Treatment of isolated perfused lung preparations with SH045 led to a decrease in lung ischemia-reperfusion edema (LIRE), a life-threatening condition associated with TRPC6 that may occur after organ transplantation. |
|
| DC70796 |
SRI-41315
Featured
|
SRI-41315 is a small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion, restores CFTR expression and function, suppresses CFTR nonsense mutations.SRI-41315 induces translational readthrough by depleting eRF1 protein level and prolonging the translational pause that occurs at premature termination codons (PTCs).SRI-41315 reduced eRF1 levels in a manner dependent upon a ubiquitin-mediated proteasome degradation pathway.SRI-41315 in combination with G418 restores CFTR function in primary bronchial epithelial cells derived from a CF patient with CFTR nonsense alleles. |

|
| DC70804 |
SSCI-1 |
SSCI-1 is a highly potent, selective NaV1.7 inhibitor with IC50 of 27 and 82 nM on human and rhesus Nav1.7 channels, respectively.SSCI-1 showed robust selectivity over other human and rhesus Nav channel paralogs, with the exception of Nav1.2 channels (IC50 252 nM for hNaV1.2), shows no other channels and receptors in 114 enzymatic, radioligand binding, and cellular assays.SSCI-1 also exhibits high potency in manual patch clamp experiments (IC50=66/295 nM for huma/rhesus Nav1.7 channels).SSCI-1 does not affect myelinated nerve excitability as measured by TT, SSCI-1 inhibits odorant-induced olfaction in the olfactory bulb of rhesus monkeys, measured by fMRI.SSCI-1 inhibits withdrawal responses to noxious heat in rhesus monkeys. |
|
| DC70805 |
SSCI-2
Featured
|
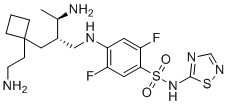
SSCI-2 is a highly potent, selective NaV1.7 inhibitor with IC50 of 9 and 17 nM on human and rhesus Nav1.7 channels, respectively.SSCI-2 showed robust selectivity over other human and rhesus Nav channel paralogs, with the exception of Nav1.2 channels (IC50 183 nM for hNaV1.2), shows no other channels and receptors in 114 enzymatic, radioligand binding, and cellular assays.SSCI-2 also exhibits high potency in manual patch clamp experiments (IC50=24/62 nM for huma/rhesus Nav1.7 channels).SSCI-2 inhibits withdrawal responses to noxious heat in rhesus monkeys. |
|
| DC70811 |
STS66 |
STS66 is a novel BBB-penetrant NKCC1 inhibitor.STS66 treatment completely blocked the ischemia-induced elevation of pNKCC1, which is superior to BMT and STS5.STS66 showed efficacy against permanent focal ischemic brain injury with hypertension comorbidity. |
|
| DC70827 |
tatM2NX |
tatM2NX is a novel potent, selective, cell permeable, peptide TRPM2 channel antagonist with IC50 of 396 nM, IC90 of 2 uM (TRPM2 channel currents), prevents ligand binding and TRPM2 activation.tatM2NX interacts with the ADPR binding site on the NUT9-H domain of TRPM2 channel. tatM2NX inhibits TRPM2-mediated dephosphorylation of GSK3β (activation), tatM2NX demonstrated selective neuroprotective effects in a mouse model of focal ischemia (stroke). |
|
| DC70830 |
TC-6683 |
TC-6683 (AZD1446) is a potent and highly selective α4β2 nAChR agonist with Ki of 30/34 nM against hα4β2/rα4β2, with little to no affinity against h α3β2 and h α7 (Ki>6.7 uM).TC-6683 (10 uM) produced <30% inhibition of specific binding or enzyme activity at all targets except the human (87%) and murine (97%) 5-HT3 receptors at a panel of 70 receptors, ion channels, and enzymes at NovaScreen/Caliper.TC-6683 (AZD1446) enhanced working memory in the object recognition paradigm in rats at every dose tested (0.1−3 mg/kg, po), exhibited favorable pharmaceutical properties and in vivo efficacy in animal models. |
|
| DC70877 |
USP5-Cav3.2 inhibitor II-1 |
USP5-Cav3.2 inhibitor II-1 is a small molecule inhibitor of USP5-Cav3.2 interaction, without affecting the enzymatic activity of USP5.II-1 inhibited both phases of formalin-induced nocifensive behaviors in mice.Cav3.2 is required for the action of II-1.II-1 also mediated a robust inhibition of mechanical allodynia induced by injury to the sciatic nerve. |
|
| DC70883 |
VK-II-86
Featured
|
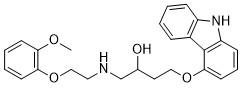
VK-II-86 is a carvedilol analogue lacking antagonist activity at β-adrenoceptors, effectively suppresses SOICR by directly reducing the open duration of the cardiac ryanodine receptor (RyR2).VK-II-86 exhibited >2,000-fold lower beta-AR binding affinity than carvedilol.VK-II-86 prevented stress-induced ventricular tachyarrhythmias in RyR2-mutant mice and did so more effectively when combined with either of the selective beta blockers metoprolol or bisoprolol.VK-II-86 prevented hypokalaemia-induced AP prolongation and depolarization but did not alter AP parameters in normokalaemia. |
|
| DC70890 |
VU0606170
Featured
|
VU 0606170 (also referred to as VU0606170) is a selective inhibitor of the sodium-activated potassium channel KNa1.1, also known as Slack or Slo2.2. This compound has shown specificity for Slack channels over other related potassium channels, making it a valuable tool for studying the physiological and pathological roles of KNa1.1. |
|
| DC70892 |
VU0468554 |
VU0468554 (VU 0468554) is a nove potent, selective inhibitor of GIRK channel with IC50 of 0.85 and 2.6 uM for GIRK1/GIRK4 and GIRK1/GIRK2 in cellular assays.VU0468554 more effectively inhibits the cardiac GIRK channel than the neuronal GIRK channel, inhibits Gβγ-activated GIRK channels in noncompetitive and potentially uncompetitive fashion.VU0468554 competitively inhibits GIRK-channel activation by ML297, a GIRK-channel activator.In the isolated heart model, VU0468554 partially reversed carbachol-induced bradycardia in hearts from wild-type mice but not Girk4-/- mice. |
|
| DC70898 |
VU6036720 |
VU6036720 (VU 6036720) is the first potent, selective inhibitor of heteromeric Kir4.1/Kir5.1 (KCNJ10/KCNJ16) with IC50 of 0.24 uM.VU6036720 displays >30-fold selective for Kir4.1/5.1 over homomeric Kir4.1 channels, Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir4.2, Kir6.2/SUR1, Kir6.1/SUR2b and Kir7.1.VU6036720 inhibits Kir4.1/5.1 activity through a reduction of channel open-state probability and single-channel current amplitude, does not induce diuresis in vivo.VU6036720 showwd an IC50 in 3 uM in Tl+ flux assays, approximately 7-times more potent at inhibiting Kir4.1/5.1 than fluoxetine and amitriptyline.VU6036720 represents the current state-of-the-art Kir4.1/5.1 inhibitor that should be useful for probing the functions of Kir4.1/5.1 in vitro and ex vivo. |
|