 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
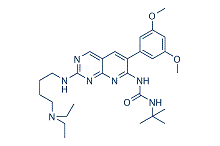




















| Cas No.: | 219580-11-7 |
| Chemical Name: | 1-tert-butyl-3-(2-(4-(diethylamino)butylamino)-6-(3,5-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl)urea |
| Synonyms: | PD173074, PD 173074 |
| SMILES: | N(C(C)(C)C)C(NC1=NC2C(C=C1C1=CC(OC)=CC(OC)=C1)=CN=C(NCCCCN(CC)CC)N=2)=O |
| Formula: | C28H41N7O3 |
| M.Wt: | 523.67 |
| Purity: | >98% |
| Sotrage: | 2 years -20°C Powder, 2 weeks 4°C in DMSO, 6 months -80°C in DMSO |
| Description: | PD173074 is a potent FGFR1 inhibitor with an IC50 of 25 nM and also inhibits VEGFR2 with an IC50 of 100-200 nM, showing 1000-fold selectivity for FGFR1 over PDGFR and c-Src. |
| Target: | FGFR1:25 nM (IC50) VEGFR2:100 nM (IC50) |
| In Vivo: | PD 173074 (1 mg/kg, i.p.) exhibits dose-dependent inhibition of FGF-induced neovascularization and angiogenesis in mice[1]. D173074 (25 mg/kg, p.o.) significantly inhibits tumor growth in mice[4]. |
| In Vitro: | PD 173074 inhibits autophosphorylation of FGFR1 in a dose-dependent manner with an IC50 in the range 1-5 nM. PD 173074 is an ATP-competitive inhibitor of FGFR1 with an inhibitory constant (Ki) of 40 nM[1]. PD 173074 and SU 5402 produce concentration-dependent reductions in FGF-2 enhancement of granule neuron survival, with IC50 values of 8 nM and 9 μM, respectively. PD 173074 does not inhibit neurotrophic and neuritogenic actions of FGF-2 signalling molecules in cerebellar granule neurons. PD 173074 and SU 5402 concentration-dependently inhibits the neurite growth response, when tested on FGF-2-treated granule neurons growing on polylysine/laminin, with IC50s of 22 nM and 25 μM, respectively[2]. PD173074 effectively antagonizes the effect of FGF-2 on proliferation and differentiation of OL progenitors in culture. Mitogen-activated protein kinase (MAPK) activation, a downstream event after activation of either FGFR or PDGFR, is also blocked by PD173074 in OL progenitors stimulated with FGF-2 but not PDGF[3]. |
| Cell Assay: | An NIH 3T3 cell line overexpressing VEGFR2 (Flk-1) has been described previously. This cell line also expresses FGFR1 endogenously. Cells (1×106) in DMEM supplemented with 10% calf serum are seeded in 10 cm2 dishes and allowed to grow for 48 h. The medium is then removed and the cells are made quiescent in starvation medium (DMEM with 0.1% calf serum). After 18 h, the cells are incubated for 5 min with various concentrations of PD 173074 prepared in starvation medium. The cells are then stimulated with growth factor [VEGF (100 ng/mL) or aFGF (100 ng/mL) and heparin (10 µg/mL)] for 5 min at 37°C. The cells are washed with ice-cold PBS and lysed in 1 mL of lysis buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EGTA, 1.5 mM MgCl2, 1 mM PMSF, 10 µg/mL aprotonin, 10 µg/mL leupeptin) containing phosphatase inhibitor (0.2 mM Na3VO4). For inhibition studies of FGFR1, cell lysates are immunoprecipitated with antibodies to FGFR1, and then analyzed by SDS-PAGE and immunoblotting with antibodies to phosphotyrosine. For inhibition studies of VEGFR2, cell lysates (20 µL) are analyzed directly by SDS-PAGE and immunoblotted with antibodies to phosphotyrosine. |
| Animal Administration: | Six-week-old athymic nude mice are inoculated subcutaneously with 3×105 NIH 3T3 cells expressing Y373C FGFR3 and Ras V12. Intraperitoneal injections of either 20 mg/kg PD173074 or 0.05 mol/Llactic acid buffer are initiated on the day of tumor injection and continued for 9 days. Ten mice for each experiment are included. |
| References: | [1]. Mohammadi M, et al. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998 Oct 15;17(20):5896-904. [2]. Skaper SD, et al. The FGFR1 inhibitor PD 173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J Neurochem. 2000 Oct;75(4):1520-7. [3]. Bansal R, et al. Specific inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation, and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J Neurosci Res. 2003 Nov 15;74(4):486-93. [4]. Trudel S, et al. Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t(4;14) myeloma. Blood. 2004 May 1;103(9):3521-8. [5]. Mahe M, et al. An FGFR3/MYC positive feedback loop provides new opportunities for targeted therapies in bladder cancers. EMBO Mol Med. 2018 Apr;10(4). pii: e8163. [6]. Zheng Y, et al. Inhibition of FGFR Signaling With PD173074 Ameliorates Monocrotaline-induced Pulmonary Arterial Hypertension and Rescues BMPR-II Expression. J Cardiovasc Pharmacol. 2015 Nov;66(5):504-14. |