 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
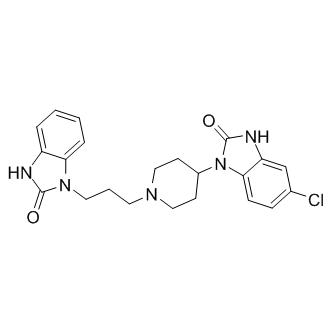
| DC9133 | Domperidone |
Domperidone is a dopamine blocker and an antidopaminergic reagent.
More description
|
|
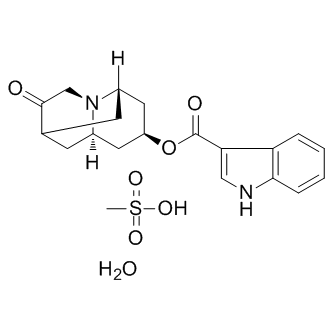
| DC9323 | Dalasetron (Mesylate hydrate) |
Dolasetron(MDL-73147) is a serotonin 5-HT3 receptor antagonist used to treat nausea and vomiting following chemotherapy.
More description
|
|
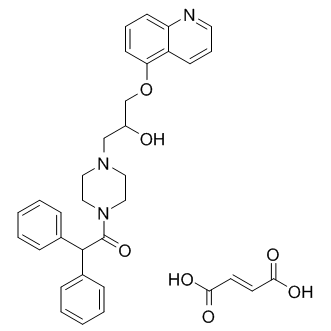
| DC9588 | Dofequidar (fumarate) |
Dofequidar fumarate(MS-209 fumarate), an orally active quinoline compound, has been reported to overcome MDR by inhibiting ABCB1/P-gp, ABCC1/MDR-associated protein 1, or both.
More description
|
|
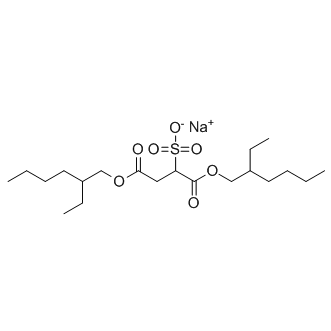
| DC10247 | Docusate Sodium |
Docusate Sodium is a laxative used to treat constipation, for constipation due to the use of opiates it maybe used with a stimulant laxative, can be taken by mouth or rectally.
More description
|
|
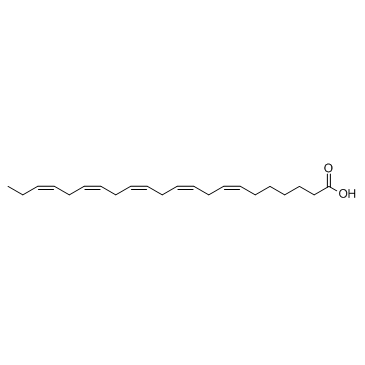
| DC12208 | Docosapentaenoic acid 22n-3 |
Docosapentaenoic acid (22n-3) is a component of phospholipids found in all animal cell membranes.
More description
|
|
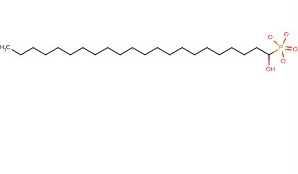
| DCAPI1299 | Docosanol (Abreva) |
Docosanol (Abreva)
More description
|
|
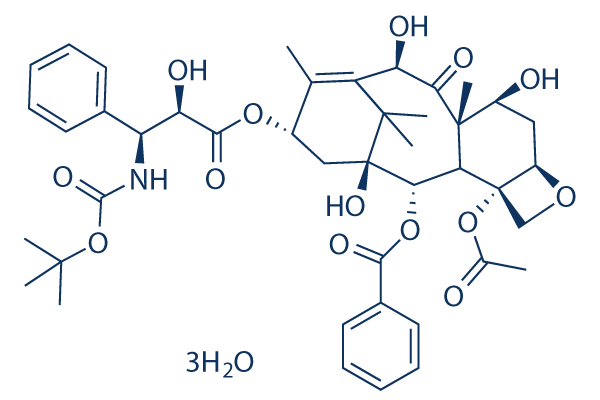
| DC8416 | Docetaxel Trihydrate |
Docetaxel, an analog of paclitaxel, is an inhibitor of depolymerisation of microtubules by binding to stabilized microtubules.
More description
|
|
| DC20960 | DO-34 |
DO-34 is a highly potent dual DAGLα/β inhibitor with IC50 of 6 nM 3-8 nM, respectively.
More description
|

|
| DC20364 | DNS-pE2 |
DNS-pE2 is the first small molecule that can selectively label endogenous 3-phosphoglycerate dehydrogenase (PHGDH) from various mammalian cells.
More description
|
|
| DC21509 | DNS-pE |
DNS-pE (PHGDH Fluorescent Probe DNS-pE) is the first small molecule that can selectively label endogenous 3-phosphoglycerate dehydrogenase (PHGDH) from various mammalian cells.
More description
|
|
| DC11345 | MMP-7 Fluorogenic Substrate |
Dnp-RPLALWRS is a fluorogenic substrate for matrix metalloproteinase-7 (MMP-7).The activity of MMP-7 can be quantified by measuring tryptophan fluorescence that is unquenched upon peptide hydrolysis that removes the N-terminal dinitrophenol (Dnp) group.
More description
|
|
| DC11344 | MMP-2/MMP-9 Fluorogenic Substrate I |
Dnp-PLGMWSR is a fluorogenic substrate for matrix metalloproteinase-2 (MMP-2) and MMP-9.The activity of MMP-2 and MMP-9 can be quantified by measuring tryptophan fluorescence that is unquenched upon peptide hydrolysis that removes the N-terminal dinitroph
More description
|
|
| DC11342 | MMP-1 Fluorogenic Substrate III |
Dnp-P-Cha-Abu-Cys(Me)-HA-K(Nma)-NH2 is a fluorogenic substrate for matrix metalloproteinase-1 (MMP-1) and MMP-9.
More description
|
|
| DC11337 | MMP Substrate II Control |
Dnp-GPLG is a peptide for use as a negative control for activity of matrix metalloproteinases (MMPs) that cleave peptides containing the sequence Dnp-GPLG.
More description
|
|
| DC23671 | DNMDP |
DNMDP is a potent and selective cancer cell cytotoxic agent that binds to PDE3A, promotes an interaction between PDE3A and Schlafen 12 (SLFN12).
More description
|
|
| DC20959 | DNDI-8219 |
DNDI-8219 is a novel potent, orally active antileishmanial agent with IC50 of 0.19 uM against Leishmania donovani (L. don).
More description
|
|
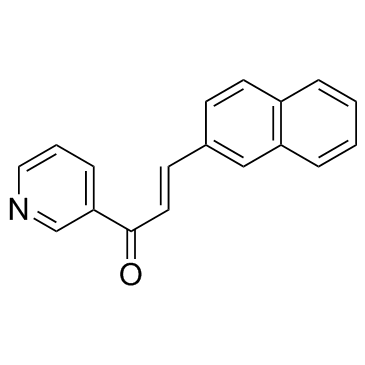
| DC12079 | DMU2105 |
DMU2105 is a potent and specific CYP1B1 inhibitor, with IC50s of 10, 742 nM for CYP1B1 and CYP1A1, respectively.
More description
|
|
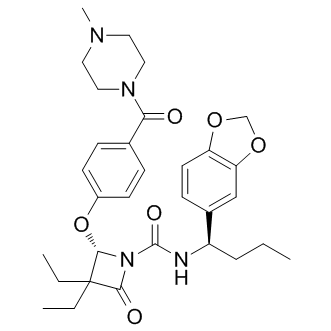
| DC9434 | DMP 777 |
DMP 777(L-694458) is a potent, selective, and orally active human leukocyte elastase (HLE) inhibitor.
More description
|
|
| DCAPI1183 | DL-Carnitine HCl |
DL-Carnitine HCl
More description
|
|
| DC20363 | DL-AP4 sodium salt |
DL-AP4 sodium salt is a broad spectrum glutamate receptor antagonist..
More description
|
|
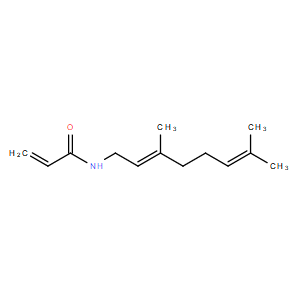
| DC11025 | DKM 3-42 |
DKM 3-42 is a selective, covalent, in vivo-active ALDH3A1 inhibitor with IC50 of 50 uM against purified human ALDH3A1, targets ALDH3A1 catalytic cysteine, impairs both in situ and in vivo lung cancer pathogenicity.
More description
|
|
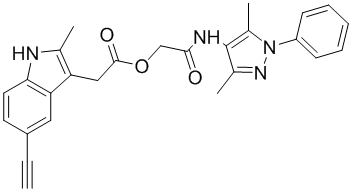
| DC12404 | DKFZ-633 |
DKFZ-633 (DKFZ633) is a selective inhibitor of Kallikrein-related peptidase 6 (KLK6) with IC50 of 0.25 uM, demonstrates good selectivity for KLK6 compared to other KLKs.
More description
|
|
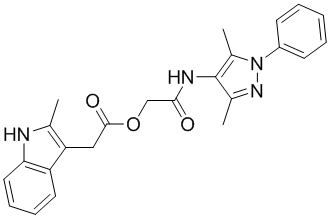
| DC12431 | DKFZ-251 |
DKFZ-251 (DKFZ251) is a selective inhibitor of Kallikrein-related peptidase 6 (KLK6) with IC50 of 0.13 uM, demonstrates good selectivity for KLK6 compared to other KLKs.
More description
|
|
| DCAPI1335 | Divalproex sodium |
Divalproex sodium
More description
|
|
| DC22511 | Disitertide |
Disitertide (P144) is a TGF-β1 antagonist peptide.
More description
|
|
| DCAPI1267 | Dipyridamole (Persantine) |
Dipyridamole (Persantine)
More description
|
|
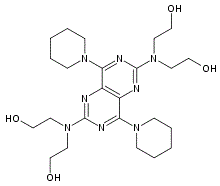
| DC11244 | Diprovocim |
Diprovocim is a potent human and mouse Toll-like receptor (TLR)1/TLR2 agonist.
More description
|
|
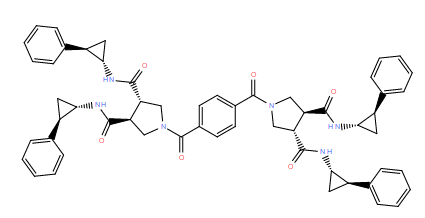
| DC12269 | Diprotin A TFA (Ile-Pro-Pro (TFA)) |
Diprotin A (TFA) is an inhibitor of dipeptidyl peptidase IV (DPP-IV).
More description
|
|
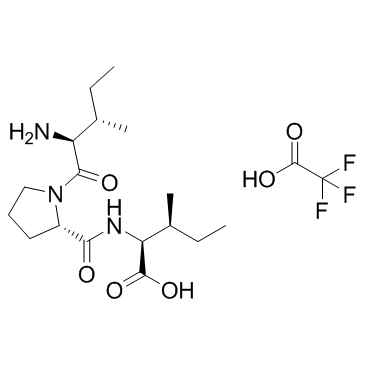
| DC11361 | Dipivefrin (hydrochloride) |
Dipivefrin is a prodrug of epinephrine that is hydrolyzed by cholinesterase and other esterases in the cornea to epinephrine.
More description
|
|
| DC23591 | Diphyllin |
Diphyllin is a naturally potent vacuolar ATPase (v-ATPase) inhibitor with IC50 of 17 nM, potently inhibits the acid influx with IC50 of 0.6 nM.
More description
|
|