 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DCAPI1582 | Diphenhydramine Citrate |
Diphenhydramine Citrate
More description
|

|
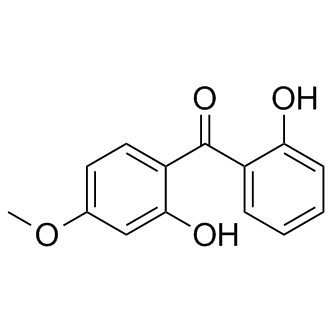
| DC10243 | 2,2′-Dihydroxy-4-methoxybenzophenone |
Dioxybenzone (benzophenone-8) is an organic compound used in sunscreen to block UVB and short-wave UVA (ultraviolet) rays
More description
|
|
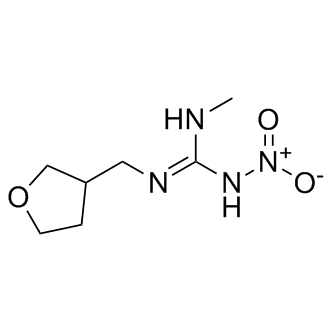
| DC8707 | Dinotefuran |
Dinotefuran is an insecticide of the neonicotinoid class, its mechanism of action involves disruption of the insect's nervous system by inhibiting nicotinic acetylcholine receptors.
More description
|
|
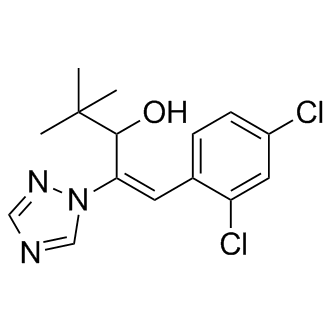
| DC8674 | Diniconazole |
Diniconazole is a newly developed fungicide strongly inhibited lanosterol 14 alpha-demethylation catalyzed by a yeast cytochrome P-450.
More description
|
|
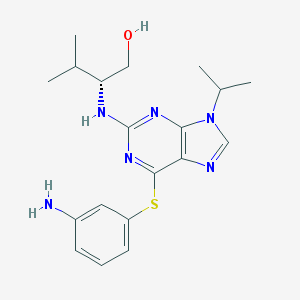
| DC8588 | Diminutol |
Diminutol is a cell-permeable 2,6,9-trisubstituted purine analog that blocks mitotic spindle assembly by competitively inhibiting NQO1, an NADP-dependent oxidoreductase.
More description
|
|
| DC23590 | Diminazene aceturate |
Diminazene aceturate is an anti-infective agent, also is a slow pore blocker of acid-sensing ion channel 1a (ASIC1a) with IC50 of 0.3 uM.
More description
|
|
| DC24169 | Dimethyl fumarate |
Dimethyl fumarate (DMF.
More description
|
|
| DC7402 | Dimethocaine |
Dimethocaine, a synthetic cocaine derivative: studies on its in vitro metabolism catalyzed by P450s and NAT2.For the detailed information of Dimethocaine, the solubility of Dimethocaine in water, the solubility of Dimethocaine in DMSO, the solubility of Dimethocaine in PBS buffer, the animal experiment (test) of Dimethocaine, the cell expriment (test) of Dimethocaine, the in vivo, in vitro and clinical trial test of Dimethocaine, the EC50, IC50,and Affinity of Dimethocaine, Please contact DC Chemicals..
More description
|

|
| DC10262 | 5,5-Dimethyloxazolidine-2,4-dione |
Dimethadione is an anticonvulsant that is the active metabolite of trimethadione.
More description
|

|
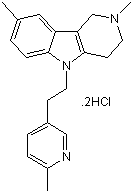
| DC4161 | Dimebon dihydrochloride |
Dimebon is an antihistamine drug.
More description
|
|
| DC20362 | DIMATE |
DIMATE is a novel irreversible, competitive, isoform-specific ALDH1 inhibitor, displays cytotoxic activity on human AML cell lines with IC50 of 1-15 uM.
More description
|
|
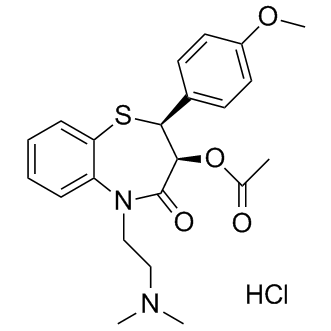
| DCAPI1152 | Diltiazem HCl (Tiazac) |
Diltiazem HCl (Tiazac)
More description
|
|
| DC11961 | Dihydromunduletone |
Dihydromunduletone is a rotenoid derivative that selectively inhibits GPR56 (IC50=21 uM) and GPR114/ADGRG5, but not GPR110 or Class A GPCRs.
More description
|

|
| DC10176 | Dihydroisotanshinone I |
Dihydroisotanshinone I is a bioactive compound present in a widely used traditional Chinese medicine named danshen.
More description
|
|
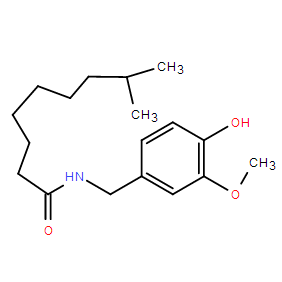
| DC23085 | Dihydrocapsaicin |
Dihydrocapsaicin, a potential inducer of autophagy, has cytotoxic activity. It has anti-atherogenic activity, can reduce the susceptibility of low-density lipoprotein (LDL) to oxidation. Dihydrocapsaicin treatment depletes peptidergic nerve fibers of subs
More description
|
|
| DC10256 | Diflunisal |
Diflunisal is a difluorophenyl derivate of salicylic acid and a nonsteroidal anti-inflammatory drug (NSAID) with antipyretic, analgesic and anti-inflammatory properties. The mechanism of action of diflunisal is as a Cyclooxygenase Inhibitor.
More description
|

|
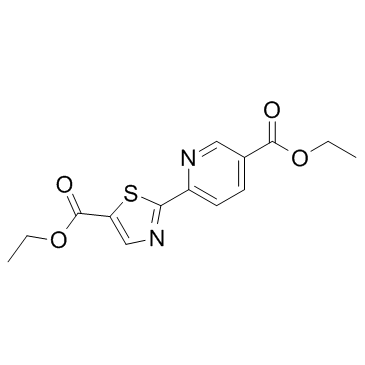
| DC12140 | Diethyl-pythiDC |
Diethyl-pythiDC is an inhibitor of collagen prolyl 4-hydroxylases (CP4Hs).
More description
|
|
| DC9890 | Diethyl maleate |
Diethylmaleate is the diethyl ester of maleic acid and a glutathione-depleting compound that inhibits NFkB.
More description
|
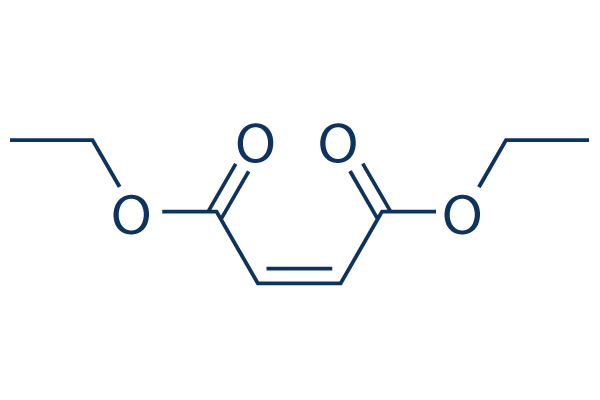
|
| DC22178 | Didemethylclomipramine |
Didemethylclomipramine (Norclomipramine, N-Desmethylcomipramine) is the major active metabolite of the tricyclic antidepressant (TCA) clomipramine.
More description
|
|
| DC9481 | Didanosine |
Didanosine(Videx) is a reverse transcriptase inhibitor with an IC50 of 0.49 μM.
Target: NRTIs; HIV
Didanosine is a dideoxynucleoside compound in which the 3'-hydroxy group on the sugar moiety has been replaced by a hydrogen.
More description
|
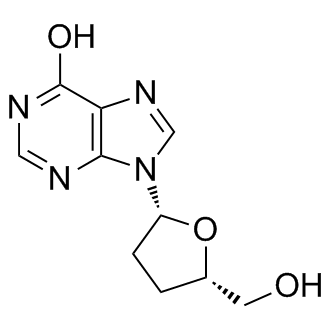
|
| DC20359 | Dicycloplatin |
Dicycloplatin (DCP) is a novel platinum analog that demonstrates significant antitumor activity against a variety of human cancer cell lines with IC50 of 25-30 nM.
More description
|
|
| DC9064 | Diclofenac sodium |
Diclofenac Sodium is a non-selective COX inhibitor with IC50 of 60 and 220 nM for ovine COX-1 and -2, respectively.
More description
|
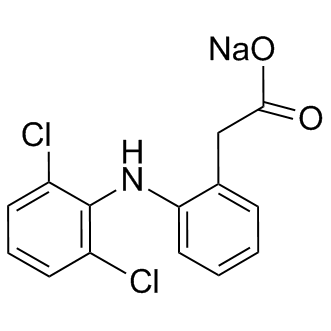
|
| DC10270 | Diazoxide |
Diazoxide is a well-known small molecule that activates KATP channels in the smooth muscle of blood vessels and pancreatic beta-cells by increasing membrane permeability to potassium ions.
More description
|

|
| DC23747 | Diazepinomicin |
Diazepinomicin (TLN 4601.
More description
|
|
| DC12077 | Diacylglycerol acyltransferase inhibitor-1 |
Diacylglycerol acyltransferase inhibitor-1 is a diacylglycerol acyltransferase (DGAT1) inhibitor.
More description
|
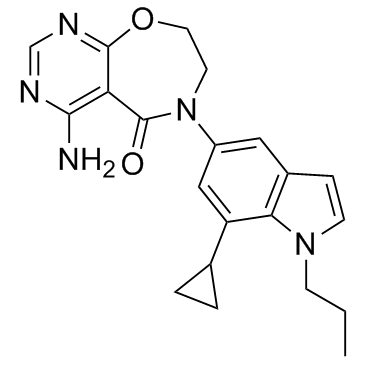
|
| DC11500 | DI-591 |
DI-591 (DI591) is a potent, selective, cell-permeable inhibitor of the DCN1-UBC12 interaction with Ki of 10-12 nM for human DCN1 and DCN2.
More description
|
|
| DC20954 | DI-404 |
DI-404 is a highly potent, peptidomimetic inhibitor of DCN1-UBC12 protein-protein interaction that bind to DCN1 protein with Kd of 6.9 nM, selectively inhibits the neddylation of cullin 3 over other cullin members.
More description
|
|
| DC20937 | D-I03 |
D-I03 (D-103) is a small molecule that inhibits RAD52-mediated ssDNA annealing with IC50 of 5 uM, binds to RAD52 with Kd of 25.8 uM and inhibits D-loop formation with IC50 of 8 uM in vitro.
More description
|
|
| DC20953 | DH-376 |
DH-376 is a highly potent dual DAGLα/β inhibitor with IC50 of 6 nM and 3-8 nM, respectively.
More description
|
|
| DCAPI1274 | D-glutamine |
D-glutamine is a D type stereoisomer of glutamine which is one of the 20 amino acids encoded by the standard genetic code.
More description
|
|