 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC23168 | CCT128930 |
CCT128930 is a potent, selective, ATP-competitive AKT inhibitor with IC50 of 6 nM (AKT2), 28-fold selectivity over the closely related PKA.
More description
|

|
| DC20872 | CCT-031374 |
CCT-031374 is a selective β-catenin signaling inhibitor with IC50 of 6.1 uM.
More description
|
|
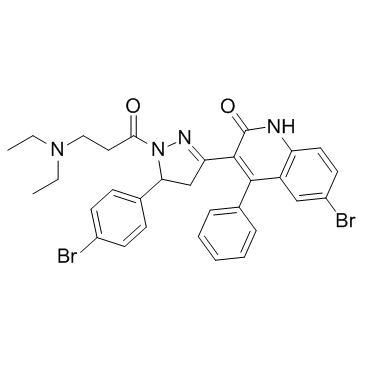
| DC12283 | CCT020312 |
CCT020312 is capable of delivering potent, and eukaryotic initiation factor 2-alpha kinase 3 (EIF2AK3) selective, proliferation control and also is an activator of RNA-like endoplasmic reticulum kinase (PERK).
More description
|
|
| DC22464 | CCT-018159 |
CCT-01815 is a novel potent inhibitor of Hsp90 ATPase activity with IC50 of 7.1 uM.
More description
|
|
| DC20874 | CCT 365623 |
CCT 365623(CCT365623, CCT-365623) is a potent, orally active small molecule inhibitor of lysyl oxidase (LOX) with IC50 of 0.89 uM.
More description
|
|
| DC20875 | CCT 365623 hydrochloride |
CCT 365623 (CCT365623) hydrochloride is a potent, orally active small molecule inhibitor of lysyl oxidase (LOX) with IC50 of 0.89 uM.
More description
|
|
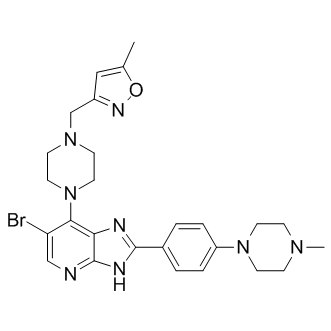
| DC8776 | CCT 137690 |
CCT 137690 is a potent inhibitor of Aurora kinases (IC50 values are 0.015, 0.019 and 0.025 μM at Aurora A, Aurora C and Aurora B respectively).
More description
|
|
| DC20876 | CCT 068127 |
CCT 068127 (CCT68127) is a novel potent inhibitor of CDK2 and CDK9 with IC50 of 10 and 90 nM for CDK2/E and CDK9/T, respectively.
More description
|
|
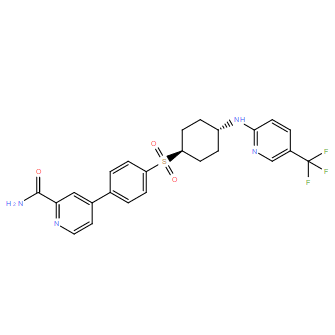
| DC11204 | CCR6 inhibitor 35 |
CCR6 inhibitor 35 is a potent and selective CCR6 inhibitor with IC50 of 6.0 nM (hCCR6).
More description
|
|
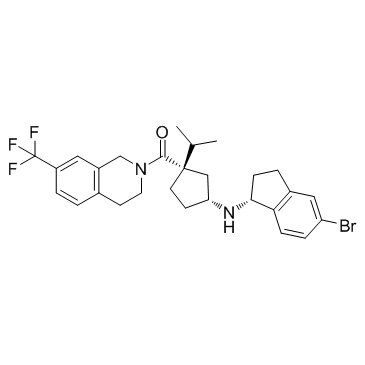
| DC12177 | CCR2 antagonist 1 |
CCR2 antagonist 1 is a high-affinity and long-residence-time CCR2 antagonist, with a Ki of 2.4 nM.
More description
|
|
| DC23528 | CCR10 antagonist 1 |
CCR10 antagonist 1 is a potent, selective CCR10 antagonist with IC50 of 690 nM.
More description
|
|
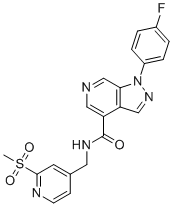
| DC12639 | CCR1 inhibitor 19e |
CCR1 inhibitor 19e is a novel potent, selective CCR1 antagonist with IC50 of 6.8 nM, inhibits CCR1chemotaxis in THP-1 cells with IC50 of 28 nM..
More description
|
|
| DC22049 | CCI-007 |
CCI-007 (CCI 007) is a selective inhibitor of MLL-r, CALM-AF10 and SET-NUP214 leukemia with IC50 of 3.5 uM against MLL-r leukemia cell line PER-485.
More description
|
|
| DC22050 | CCI-006 |
CCI-006 (CCI 006) is a novel inhibitor of MLL-rearranged and CALM-AF10 translocated leukemias that share common leukemogenic pathways.
More description
|
|
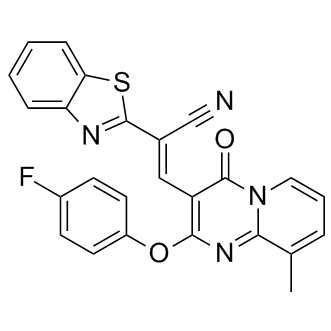
| DC9613 | CCG-63808 |
CCG-63808 is a reversible inhibitor of regulator of G-protein signaling (RGS) proteins.
More description
|
|
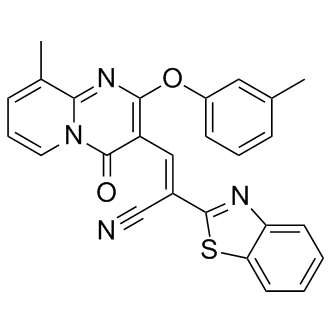
| DC9612 | CCG-63802 |
CCG-63802 is a reversible inhibitor of regulator of G-protein signaling (RGS) protein; with greatest potency at RGS4.
IC50 value:
Target: RGS
CCG-63802 is selective amongst RGS proteins, with greatest potency at RGS4.
More description
|
|
| DC20869 | CCG-232601 |
CCG-232601 is a potent (IC50=0.55 uM), orally bioavailable Rho/MKL1/SRF transcriptional pathway that target transcriptional factor MRTF.
More description
|
|
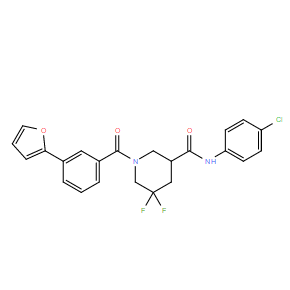
| DC10977 | CCG-222740 |
CCG-222740 (CCG222740) is a pharmacological inhibitor of MRTF/SRF signalling, shows greater inhibitory effect on MRTF/SRF target genes than MRTF-A inhibitor CCG-203971, with IC50 of 5 uM in fibroblast-mediated collagen contraction assay.
More description
|
|
| DC20871 | CCG 2979 |
CCG 2979 is a small molecule inhibitor of gene expression of streptokinase (SK), a critical group A streptococcal (GAS) virulence factor.
More description
|
|
| DC20867 | CCG 102487 |
CCG 102487 is a CCG-2979 analog that reduces streptokinase (SK) activity with little inhibition of group A streptococcal (GAS) growth.
More description
|
|
| DC23540 | CCC07-01 |
CCC07-01 (CCC 0701) is a novel alkylating compound that is capable of disrupting expression of PD-1, PD-L1 and CTLA-4 genes with IC50 of 15, 10 and 5 nM in cell-base assays.
More description
|
|
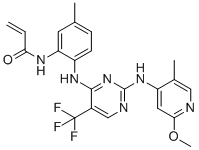
| DC12532 | CC-90003 |
CC-90003 (CC90003) is a potent and selective, covalent ERK1/2 inhibitor with IC50 of 10-20 nM, demonstrates excellent selectivity in a 258-kinase biochemical assay panel.
More description
|
|
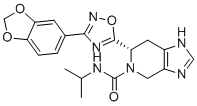
| DC12623 | CBK289001 |
CBK289001 (CBK-289001) is a small molecule inhibitor of Tartrate-resistant acid phosphatase (TRAP/ACP5), demonstrates efficacy in a migration assay and IC50 values from 4 to 125 uM..
More description
|
|
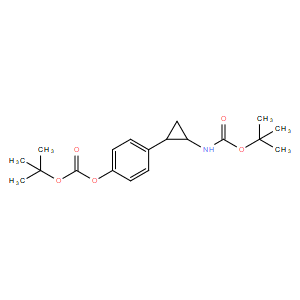
| DC10944 | CBB3001 |
CBB3001 (CBB-3001) is a novel potent, selective histone demethylase LSD1 inhibitor with IC50 of 21.25 uM.
More description
|
|
| DC20863 | CB-6673567 |
CB-6673567 is a potent, selective Adenylyl Cyclase 1 (AC1) inhibitor with IC50 of 77 uM, displays a 2- to 4-fold isoform selectivity over AC6.
More description
|
|
| DC20862 | CB-618 sodium salt |
CB-618 (CB 238618) is a novel non-β-lactam β-lactamase inhibitor that potentiates meropenem MIC values against clinical Enterobacteriaceae isolates.
More description
|
|
| DC20861 | CB-618 |
CB-618 (CB 238618) is a novel non-β-lactam β-lactamase inhibitor that potentiates meropenem MIC values against clinical Enterobacteriaceae isolates.
More description
|
|
| DC22986 | CB5305630 |
CB5305630 is a potent and noncompetitive IRE1 RNase inhibitor with IC50 of 60 nM in the FRET-derepression assay.
More description
|
|
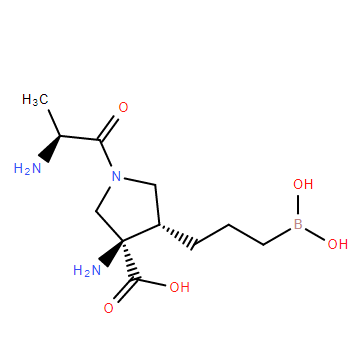
| DC11709 | CB-1158 |
CB-1158 (INCB-01158) is a potent, selective, oral available Arginase inhibitor with IC50 of 98 nM.
More description
|
|
| DC20607 | Cazpaullone |
Cazpaullone (9-Cyano-1-azapaullone) is a derivative of 1-Azakenpaullone, a potent, selective GSK3β inhibitor with IC50 of 8 nM, weakly inhibits CDK1/cyclin B and CDK5/p25 (IC50=0.5 and 0.3 uM).
More description
|
|