 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
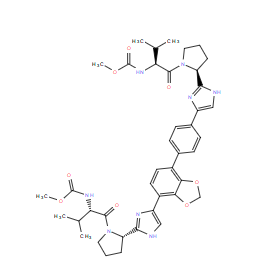
| DC11081 | Coblopasvir |
Coblopasvir (KW136, KW-136) is a novel HCV NS5A inhibitor under development for treatment of HCV infection..
More description
|
|
| DC22505 | Cobimetinib R-enantiomer |
Cobimetinib R-enantiomer (GDC-0973.
More description
|

|
| DC24119 | Cobimetinib racemate |
Cobimetinib racemate is the racemate form of Cobimetinib (GDC-0973, XL518), which is a potent, highly selective inhibitor of MEK1/2..
More description
|
|
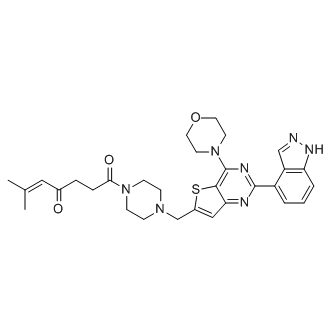
| DC7104 | CNX-1351 |
CNX-1351 is a selective covalent Inhibitor of PI3Kα. CNX-1351 was tested against all four of the class I PI3K enzymes α, β, γ, and δ.
More description
|
|
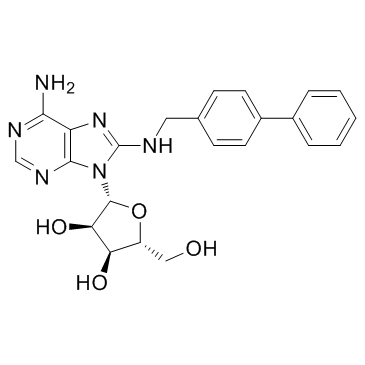
| DC12103 | CNT2 inhibitor-1 |
CNT2 inhibitor-1 is a potent concentrative nucleoside transporter 2 Inhibitor (CNT2), with an IC50 of 640 nM for hCNT2.
More description
|
|
| DC20346 | CN-SAH |
CN-SAH is a potent and selective inhibitor of histone methyl transferase DOT1L with IC50 of 26 nM, displays >10-fold selectivity over DNMT1, PRMT3, PRMT5, and G9α..
More description
|
|
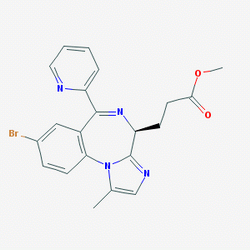
| DC7009 | CNS-7056 |
CNS 7056 is a new short-acting sedative/anaesthetic that acts on GABAA receptors in the brain.
More description
|
|
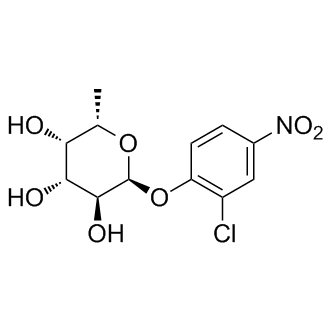
| DC9370 | CNP-AFU |
CNP-AFU (2-Chloro-4-nitrophenyl α-L-fucopyranoside) is a substrate for alpha-L-fucosidase(AFU).
More description
|
|
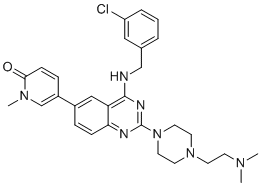
| DC12477 | CN750 |
CN750 (CN-750) is novel BRD4 (BD1, 2) inhibitor with IC50 of 44 nM, MV4-11 IC50 of 0.54 uM.
More description
|
|
| DC23508 | CMPD167 |
CMPD167 (MRK-1) is an orally acitve small molecule that binds to CCR5 to inhibit gp120 association, inhibits different stages of the virus-cell attachment and entry process (CCR5-using virus SHIV-162P3, IC50<1 nM)..
More description
|
|
| DC21531 | CMP-5 hydrochloride |
CMP-5 (PRMT5-IN-5)is a first-in-class, small-molecule PRMT5-specific inhibitor that blocks EBV-driven B-lymphocyte transformation and survival, without effect on normal B cells.
More description
|
|
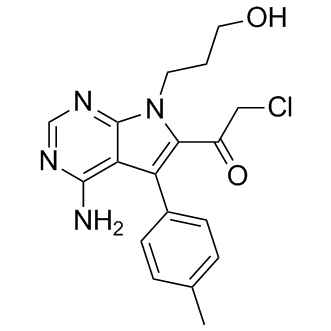
| DC9617 | CMK |
CMK is a RSK2 kinase inhibitor.
More description
|
|
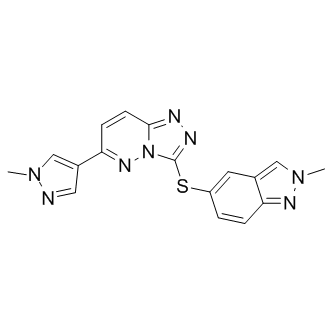
| DC7388 | c-Met inhibitor 1 |
c-Met inhibitor 1 is an inhibitor of the c-Met receptor signaling pathway useful for the treatment of cancer including gastric, glioblastoma, and pancreatic cancer.
More description
|
|
| DC23813 | CMC2.24 |
CMC2.24 (TRB-N0224) is a novel tricarbonylmethane agent that inhibits the Ras-RAF-MEK-ERK pathway, inhibits Ras activation in pancreatic cancer both in vitro and in vivo.
More description
|
|
| DCAPI1542 | CMC·HCI |
CMC·HCI
More description
|
|
| DC20913 | CMA-008 |
CMA-008 is a cell-permeable, small molecule modulator of NAADP-mediated Ca2+ release, competes with NAADP binding, inhibit Ca2+ release via the NAADP receptor (IC50=15 uM-1 mM).
More description
|
|
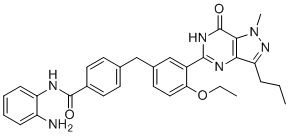
| DC12629 | CM-675 |
CM-675 (CM675) is a potent, dual PDE5 and classI-HDAC inhibitor with IC50 of 114/673 nM for PDE5A/HDAC1, respectively.
More description
|
|
| DC21063 | CM03 |
CM03 is a G-quadruplex-binding compound that potently inhibits cell growth in PDAC cell lines (MIA PaCa-2 IC50=19 nM).
More description
|
|
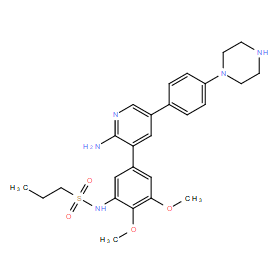
| DC11068 | CLSP43 |
CLSP43 (RIPK2 inhibitor CLSP43) is a novel potent, ATP pocket-binding RIPK2 inhibitor with IC50 of 19.9 nM in ADPGlo assay, demonstrates inhibition of both the RIPK2-XIAP interaction, and of cellular and in vivo NOD2 signaling.
More description
|
|
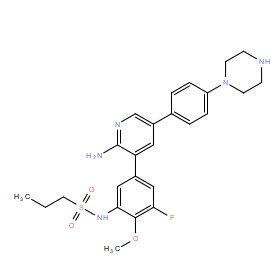
| DC11067 | CLSP37 |
CLSP37 (RIPK2 inhibitor CLSP37) is a novel potent, ATP pocket-binding RIPK2 inhibitor with IC50 of 16.3 nM in ADPGlo assay, demonstrates inhibition of both the RIPK2-XIAP interaction, and of cellular and in vivo NOD2 signaling.
More description
|
|
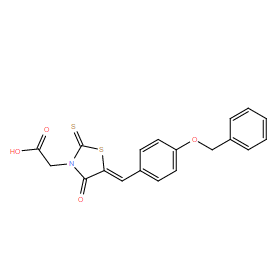
| DC11061 | ClpP inhibitor M21 |
ClpP inhibitor M21 is a noncompetitive inhibitor of ClpP with IC50 of 41.42 uM, inhibits ClpP cleavage of SUc-LY-AMC substrate in a peptidase assay.
More description
|
|
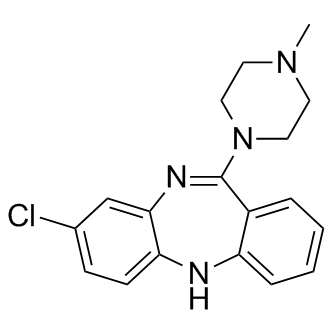
| DC9067 | Clozapine |
Clozapine(HF 1854) is a 5-HT2A/2C and dopamine receptor blocker with Ki values of 21, 170, 170, 230 and 330 nM for D4, D3, D1, D2 and D5 receptors respectively.
More description
|
|
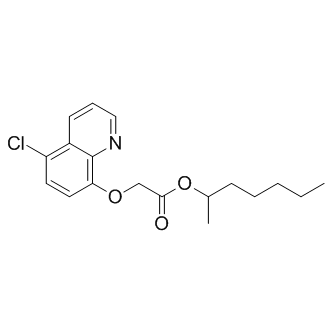
| DC8672 | Cloquintocet-mexyl |
Cloquintocet-mexyl is a herbicide, used to control coarse annual grass of the family poaceae (gramineae).
More description
|
|
| DCAPI1545 | Cloprostenol sodium |
Cloprostenol Sodium is a synthetic F series prostaglandin that functions as a potent agonist towards the PGF2αR (FP receptor). It is known that Cloprostenol Sodium is less selective than other agonists such as fluprostenol, however it displays more potenc
More description
|
|
| DCAPI1417 | Clopidogrel |
Clopidogrel is an irreversible inhibitor of the P2Y12 receptor, which is responsible for initiating signal transduction via a GPCR. The signal cascade initiates platelet aggregation and thus clopidogrel has the effect of “thinning” the blood.
More description
|
|
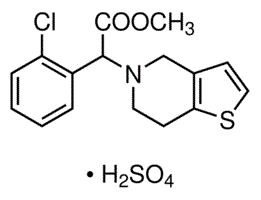
| DC3147 | Clopidogrel hydrogensulfate |
Clopidogrel (Plavix) is an oral, thienopyridine class antiplatelet agent.
More description
|
|
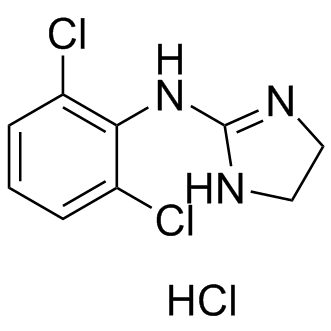
| DC9021 | Clonidine hydrochloride |
Clonidine Hydrochloride is a centrally acting alpha-agonist hypotensive agent.
More description
|
|
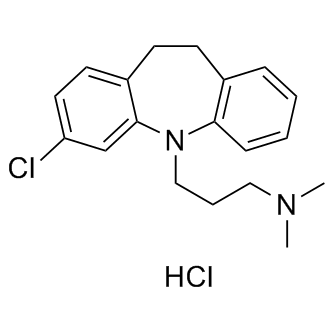
| DC9081 | Clomipramine HCL |
Clomipramine HCl is a serotonin transporter (SERT), norepinephrine transporter (NET) dopamine transporter (DAT) blocker with Ki of 0.14, 54 and 3 nM, respectively.
More description
|
|
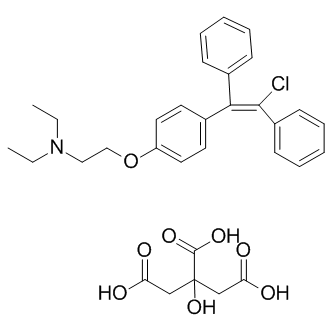
| DC9183 | Clomiphene citrate |
Clomifene Citrate is a selective estrogen receptor modulator.
More description
|
|
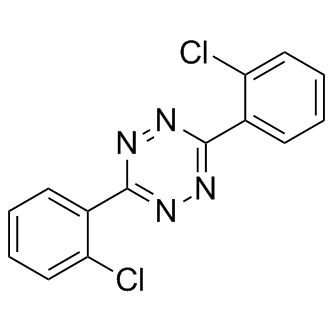
| DC8681 | Clofentezine |
Clofentezine, a growth inhibitor, has sublethal effects on life-table parameters of Tetranychus urticae Koch females.
More description
|
|