 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
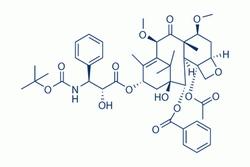
| DC4186 | Cabazitaxel |
Cabazitaxel (Jevtana, XRP6258) is a semi-synthetic derivative of a natural taxoid.
More description
|
|
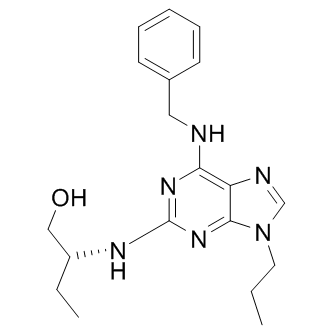
| DC8704 | Ca2+ channel agonist 1 |
Ca2+ channel agonist 1 is a N-type Ca2+ channel activity agonist, with EC50 of 14.23 uM, also inhibits cdk2 kinase activity with EC50 of 3.34 uM.
More description
|
|
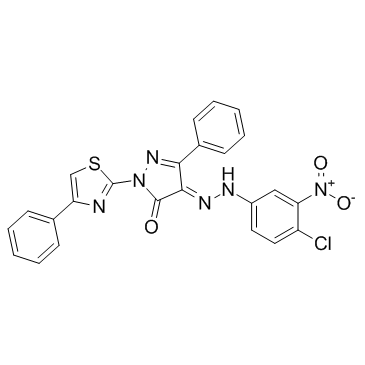
| DC10419 | C 87 |
C87 is a novel small-molecule TNFα inhibitor; potently inhibits TNFα-induced cytotoxicity with an IC50 of 8.73 μM.
More description
|
|
| DC20328 | C5-benzyl SAHA |
C5-benzyl SAHA is a C5-modified SAHA analog that displays dual selectivity to HDAC6 (IC50=0.27 uM) and HDAC8 (IC50=0.38 uM) over HDAC 1, 2, and 3 (IC50=2.9-5.8 uM).
More description
|

|
| DC20327 | C450-0730 |
C450-0730 is a potent LuxN antagonist that antagonizes LuxN-AI-1 binding with IC50 of 2.7 uM.
More description
|
|
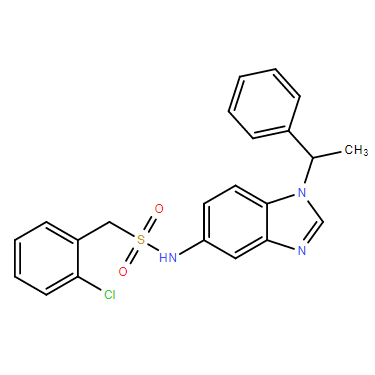
| DC11493 | C2BA-4 |
C2BA-4(C2BA 4) is a potent, specific human Pregnane X receptor (hPXR) activator with EC50 of 49 nM.
More description
|
|
| DC20855 | C-215 |
C-215 is a novel inhibitor of mycobacterial membrane protein large 3 (MmpL3) with MIC90 of 16 uM against M. tuberculosis..
More description
|
|
| DC23414 | C-021 |
C-021 is a potent and orally bioavailable CCR4 antagonist with IC50 of 0.14 uM and 0.039 uM for inhibition of chemotaxis in human and mouse, respectively..
More description
|
|
| DC23462 | C-021 dihydrochloride |
C-021 dihydrochloride is a potent and orally bioavailable CCR4 antagonist with IC50 of 0.14 uM and 0.039 uM for inhibition of chemotaxis in human and mouse, respectively..
More description
|
|
| DC20326 | BzDANP |
BzDANP is a small molecule modulator of pre-miR-29a maturation by Dicer, effectively stabilizes the C-bulged RNA and suppresses Dicer-mediated processing of pre-miR-29a in a concentration dependent manner..
More description
|
|
| DC20853 | BZ-194 |
BZ-194 is a small molecule inhibitor of NAADP action that inhibit NAADP-mediated Ca2+ signaling in T cells, neither interferes with Ca2+ mobilization by IP3 or cyclic ADP-ribose nor with capacitative Ca2+ entry.
More description
|
|
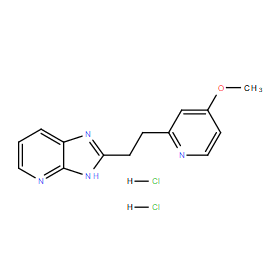
| DC11059 | BYK191023 dihydrochloride |
BYK191023 dihydrochloride (BYK-191023) is a potent, highly selective inhibitor of inducible nitric-oxide synthase (iNOS) with IC50 of 86 nM, >20-fold selectivity over nNOS and eNOS (IC50=17 and 162 uM).
More description
|
|
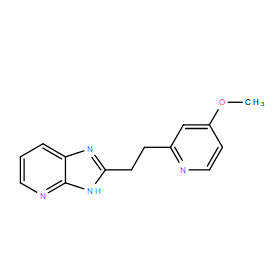
| DC11058 | BYK191023 |
BYK191023 (BYK-191023) is a potent, highly selective inhibitor of inducible nitric-oxide synthase (iNOS) with IC50 of 86 nM, >20-fold selectivity over nNOS and eNOS (IC50=17 and 162 uM).
More description
|
|
| DC22424 | BX-667 |
BX-667 is a potent P2Y12 receptor antagonist that blocks ADP-induced platelet aggregation in human, dog and rat blood (IC50=97, 317 and 3000 nM respectively).
More description
|
|
| DC20852 | BX-320 |
BX-320 is a potent, selective and ATP-competitive inhibitor of PDK1 with IC50 of 30 nM, >20-fold selectivity over PKA.
More description
|
|
| DCAPI1407 | Butenafine |
Butenafine
More description
|
|
| DC8995 | Busulfan/Myleran |
Busulfan is a bifunctional alkylating agent.
More description
|
|
| DC22437 | Burimamide oxalate |
Burimamide oxalate is a potent dual H3/H2 receptor antagonist with Ki of 0.07 and 7.8 uM, respectively.
More description
|
|
| DCAPI1459 | Bupivacaine HCL |
Bupivacaine HCL
More description
|
|
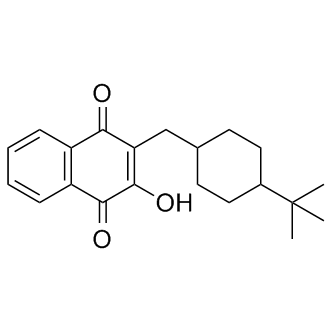
| DC9360 | Buparvaquone |
Buparvaquone(Butalex) is a hydroxynaphthoquinone antiprotozoal drug related to parvaquone and atovaquone; it is a promising compound for the therapy and prophylaxis of all forms of theileriosis.
More description
|
|
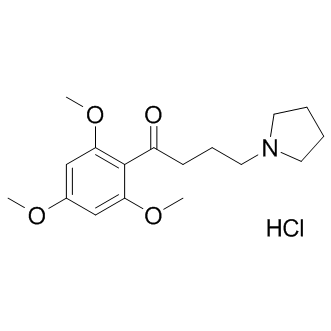
| DCAPI1160 | Buflomedil HCl |
Buflomedil HCl
More description
|
|
| DCAPI1209 | Budesonide |
Budesonide
More description
|
|
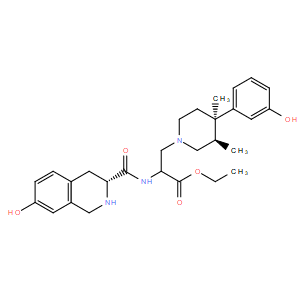
| DC10945 | BU09059 |
BU09059 is a potent, selective, short-acting kappa-opioid receptor antagonist with Ki of 1.72 nM, displays 15- and 616-fold selectivity over μ- and δ-receptors respectively.
More description
|
|
| DC22043 | BTZO-2 |
BTZO-2 is an active BTZO-1 derivative, an antioxidant response element-activator, provides protection against lethal endotoxic shock in mice..
More description
|
|
| DC22042 | BTZO-15 |
BTZO-15 is an active BTZO-1 derivative for ARE-activation with a favorable ADME-Tox profile, induces expression of heme oxygenase-1 (HO-1) and inhibits NO-induced cell death in IEC-18 cells.
More description
|
|
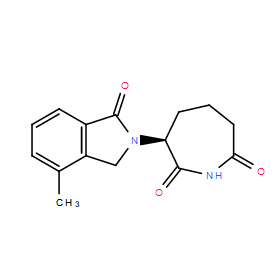
| DC11252 | BTX161 |
BTX161 is a thalidomide analog that mediates degradation of CKIα better than lenalidomide in human AML cells and activates DDR and p53, while stabilizing the p53 antagonist MDM2.
More description
|
|
| DC12457 | BTT-369 |
BTT-369 (BTT369) is a voltage-gated CaV2.2 calcium channel inhibitor that disrupts the CaVα·CaVβ3 protein-protein interaction with Ki of 2.0 uM in FP assays.
More description
|
|
| DC23870 | BTK-IN-23 |
BTK-IN-23 is a highly potent and selective Btk inhibitor with IC50 of 3 nM.
More description
|
|
| DC11431 | BTK-030 |
BTK-030 is a novel BTK inhibitor.
More description
|
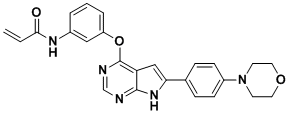
|
| DC12432 | BTK inhibitor 4b |
BTK inhibitor 4b is a potent, highly selective inhibitors of BTK with IC50 of 4.2 and 0.9 nM against activated and unactivated BTK, respectively.
More description
|
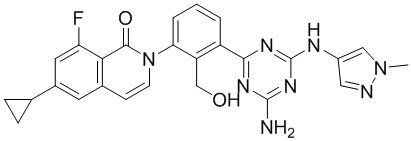
|