 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
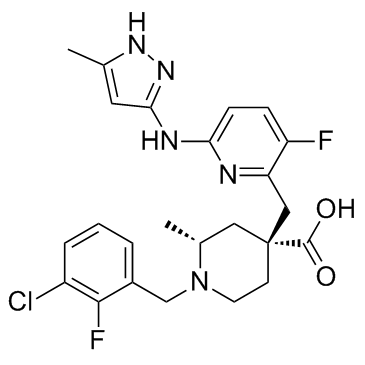
| DC12261 | LY3295668 (AK-01) |
LY3295668 is a potent, orally active and highly specific Aurora-A kinase inhibitor, with Ki values of 0.8 nM and 1038 nM for AurA and AurB, respectively.
More description
|
|
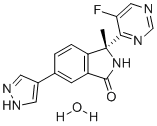
| DC12507 | LY3143921 |
LY3143921 (LY-3143921) is an orally available, potent, selective inhibitor of Cdc7 kinase inhibitor, inhibits CDC7/DBF4 I and pMCM2 (S53) with IC50 values of 3.3 nM and 290 nM, respectively..
More description
|
|
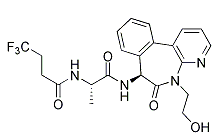
| DC7776 | LY-3039478 |
LY3039478 a orally bioavailable, novel small molecule Notch inhibitor with an IC50 of ~1nM in most of the tumor cell lines tested.
More description
|
|
| DC21340 | LY 2979165 |
LY2979165 (MP-101) is the alanine prodrug of LY2812223, a selective orthosteric mGlu2 receptor agonist.
More description
|

|
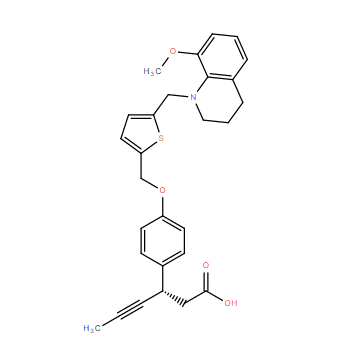
| DC11681 | LY2922470 |
LY2922470 (LY-2922470) is a potent, selective, orally available GPR40 agonist with EC50 of 7 nM.
More description
|
|
| DC22595 | LY2857785 |
LY2857785 is a potenrt, reversible and ATP-competitive CDK9 inhibitor with IC50 of 11 nM, also inhibits CDK8 (IC50=16 nM) and weakly inhibits CSK7 (IC50=246 nM).
More description
|
|
| DC23444 | LY2624803 |
LY2624803 is a novel potent histamine H1 and 5HT-2A receptor modulator in the pipeline for treating insomnia..
More description
|
|
| DC22140 | LY2623091 |
LY2623091 (LY-2623091) is an orally administered mineralocorticoid receptor antagonist for the treatment of essential hypertension and chronic kidney disease..
More description
|
|
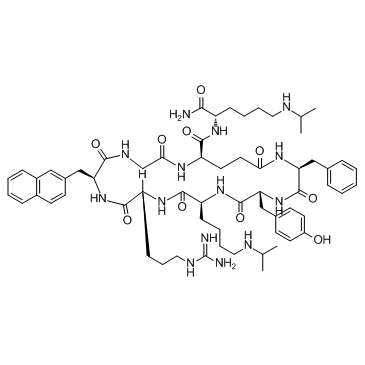
| DC10782 | LY2510924 Featured |
LY2510924 is an inhibitor of CXC chemokine receptor 4 (CXCR4), with potential antineoplastic activity.
More description
|
|
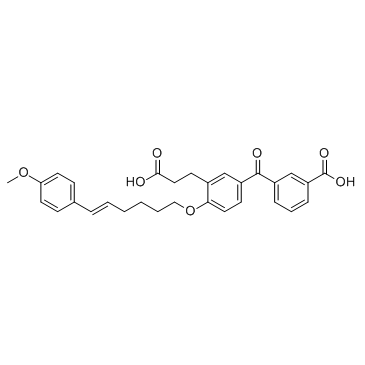
| DC12130 | LY223982 |
LY223982 is a potent and specific inhibitor of leukotriene B4 receptor, with an IC50 of 13.2 nM against [3H]LTB4 binding to LTB4 receptor.
More description
|
|
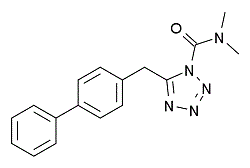
| DC2045 | LY2183240 |
LY2183240 is a novel and highly potent blocker of anandamide uptake (IC50 = 270 pM).
More description
|
|
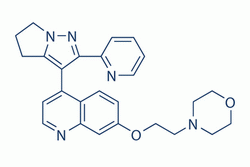
| DC7189 | LY-2109761 |
LY2109761 is a novel selective TGF-β receptor type I/II (TβRI/II) dual inhibitor with Ki of 38 nM and 300 nM, respectively; shown to negatively affect the phosphorylation of Smad2.
More description
|
|
| DC23633 | LY 392098 |
LY 392098 is a selective, potent and centrally active positive allosteric modulator of AMPAR.
More description
|
|
| DC21256 | LY 2318912 |
LY 2318912 is a potent, competitive, small molecule inhibitor of anandamide uptake with IC50 of 7.27 nM, shows high affinity against anandamide transporter binding site with Kd o f7.62 nM.
More description
|
|
| DC23136 | LX7101 |
LX7101 is a potent, dual LIMK and ROCK inhibitor with IC50 of 4.3/32//69/32/ nM for LIMK2/LIMK1/ROCK1/ROCK2, respectively.
More description
|
|
| DC21251 | LX-2931 |
LX-2931 (LX2931) is a potent, selective and orally bioavailable S1P lyase (S1PL) inhibitor.
More description
|
|
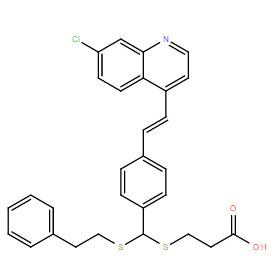
| DC11172 | LV-320 |
LV-320 (LV320) is a novel potent, cell-active, allosteric inhibitor of the autophagy-related cysteine protease ATG4B with IC50 of 24.5 uM in ATG4B cleavage assays.
More description
|
|
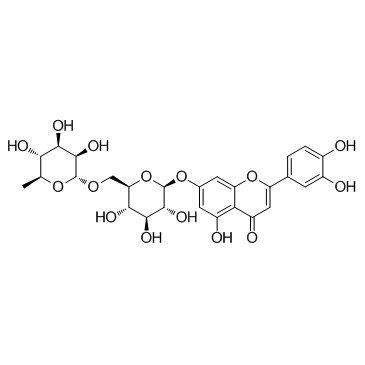
| DC12120 | Luteolin-7-rutinoside |
Luteolin-7-rutinoside has both anti-arthritic and antifungal activities, can result in a combination therapy for the treatment of fungal arthritis due to C. albicans infection.
More description
|
|
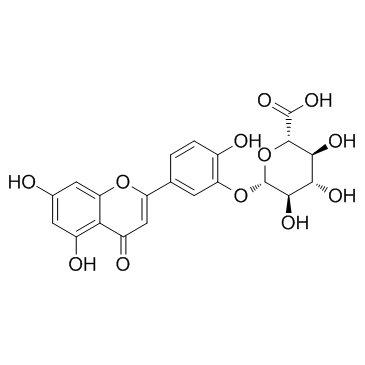
| DC12118 | Luteolin-3-O-beta-D-glucuronide |
Luteolin-3-O-beta-D-glucuronide is a luteolin glucosiduronic acid consisting of luteolin having a beta-D-glucosiduronic acid residue attached at the 3'-position.
More description
|
|
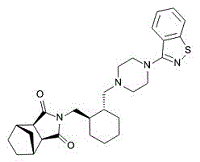
| DC3174 | lurasidone |
Lurasidone is a novel psychotropic agent that has been shown in studies of cloned human receptors to be an antagonist at the 5-HT2A receptor.
More description
|
|
| DCAPI1570 | Hydroxychloroquine Sulphate |
Lupus erythematosus suppressant as well as an antimalerial and antirheumatic.
More description
|
|
| DC22596 | LUF6000 |
LUF6000 is a potent, selective, positive allosteric modulator (enhancer) of human A3 adenosine receptor, enhance Emax but without affecting agonist potency.
More description
|
|
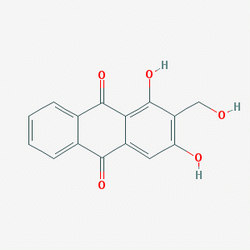
| DC7170 | Lucidin (NSC 30546) |
Lucidin (NSC 30546) is a natural component of Rubia tinctorum L. lucidin is mutagenic in bacteria and mammalian cells.
More description
|
|
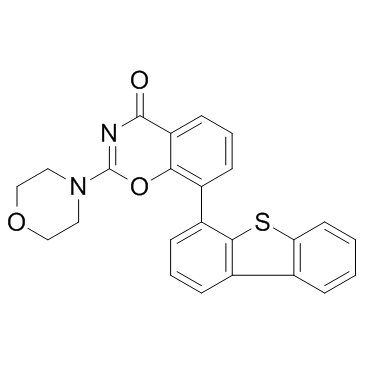
| DC24031 | LTV-1 |
LTV-1 is a highly potent, cell-permeable and reversible LYP (lymphoid tyrosine phosphatase) inhibitor with IC50 of 508 nM.
More description
|
|
| DC10155 | LTURM34 |
LTURM34 is a specific DNA-PK inhibitor with an IC50 of 0.034 μM.
More description
|
|
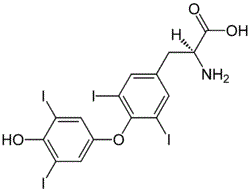
| DCAPI1211 | L-Thyroxine |
L-Thyroxine
More description
|
|
| DC23503 | LSP1-2111 |
LSP1-2111 is a potent, selective, and brain penetrant group III mGluRs agonist with EC50 of 2.2 and 1.7 uM for mGluR4 and mGluR6, respectively.
More description
|
|
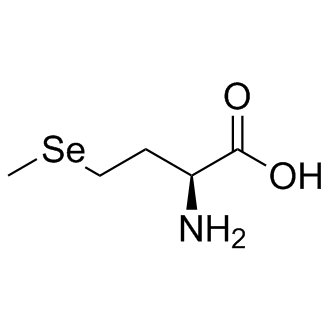
| DC8698 | L-SelenoMethionine |
L-SelenoMethionine is a major natural food-form of selenium.
More description
|
|
| DC23379 | LSD1-IN-32 |
LSD1-IN-32 is a potent, reversible lysine specific demethylase 1 (LSD1) inhibitor with biochemical IC50 of 83 nM, Kd of 32 nM, cell EC50 of 0.67 uM.
More description
|
|
| DC23384 | LSD1-IN-11p |
LSD1-IN-11p is a reversible LSD1 inhibitor with IC50/Kd of 79/21 nM.
More description
|
|