 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC21269 | Marbostat-100 |
Marbostat-100 is a potent, selective HDAC6 inhibitor with Ki of 0.7 nM, displays >200-fold selectivity over HDAC1-5, and HDAC7-10.
More description
|

|
| DC21857 | Mapracorat |
Mapracorat (ZK 245186) is a potent, selective and non-steroid glucocorticoid receptor (GR) agonist with binding Ki of 1.9 nM.
More description
|
|
| DC20442 | MAP4K4-IN-44 |
MAP4K4-IN-44 is a potent, moderately selective small molecule MAP4K4 inhibitor with IC50 of 5 nM in LC3K assay, demonstrates favorable in vivo bioavailability in mouse..
More description
|
|
| DC21916 | MAP4K4-IN-37 |
MAP4K4-IN-37 is a potent, selective, orally active inhibitor of serine-threonine kinase MAP4K4 with IC50 of 0.4 nM.
More description
|
|
| DC11315 | Manidipine(CV-4093) |
Manidipine is a dihydropyridine L- and T-type calcium channel blocker.
More description
|
|
| DC12138 | Maltohexaose (Amylohexaose) |
Maltohexaose is a natural saccharide, and can be produced from amylose, amylopectin and whole starch.
More description
|

|
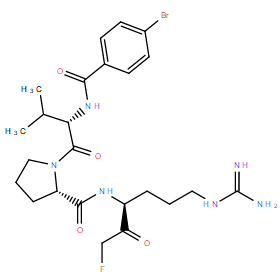
| DC11066 | MALT1 paracaspase inhibitor 3 |
MALT1 paracaspase inhibitor 3 is a potent, specific, covalent inhibitor of MALT1 paracaspase with Ki of 10 nM, exhibits 10-and 100-fold improved potency in vitro and in vivo compared with Z-VRPR-fmk.
More description
|
|
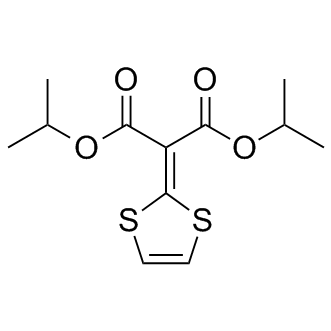
| DCAPI1277 | Malotilate |
Malotilate
More description
|
|
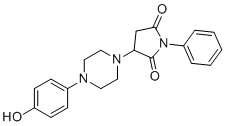
| DC12536 | Malic Enzyme inhibitor ME1 Featured |
Malic Enzyme inhibitor ME1 (ME1) is a small molecule inhibitor of Malic Enzyme (ME1) with IC50 of 0.15 uM, suppresses growth of human CRC cells in vitro, with little effect on normal rat intestinal epithelial cells..
More description
|
|
| DC23792 | MAL3-101 |
MAL3-101 is a Hsc70 modulator that inhibits Hsp70 ATPase activity, exhibits antiproliferative activity in breast cancer cells SK-BR-3 with IC50 of 27 uM.
More description
|
|
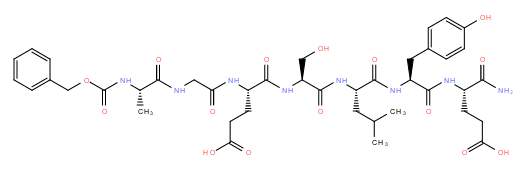
| DC11201 | MAI-400 |
MAI-400 (MAI400) is a novel peptide inhibitor of APC-Asef interaction with Kd of 12 nM and IC50 of 0.25 uM.
More description
|
|
| DC11215 | MAI-150 |
MAI-150 is a peptidomimetic inhibitor of APC-Asef interaction, blocks colorectal cancer migration..
More description
|
|
| DC9922 | Macranthoidin B |
Macranthoidin B is a major bioactive saponin in rat plasma after oral administration of extraction of saponins from Flos Lonicerae.
More description
|
|
| DC21268 | MAC173979 |
MAC173979 is a novel time-dependent inhibitor of p-aminobenzoic acid biosynthesis with IC50 of 30 uM, represents the first PABA biosynthesis inhibitor with activity against Gram-negative bacteria..
More description
|
|
| DC21267 | MAC168425 |
MAC168425 is a novel antibacterial inhibitor that interferes with glycine metabolism in E coli..
More description
|
|
| DC21266 | MAC13772 |
MAC13772 is a potent inhibitor of BioA with an IC50 of 250 nM, a novel antibacterial inhibitor that selectively inhibits PABA biosynthesis in M tuberculosis..
More description
|
|
| DC21339 | MAC1 |
MAC1 (Monastrol Antagonizing Compound 1) is a small molecule that can rescue spindle bipolarity in cells lacking Eg5 activity.
More description
|
|
| DC21265 | M-525 |
M-525 is a first-in-class, highly potent, irreversible small-molecule inhibitor of the menin-MLL interaction with binding IC50 of 3.3 nM in FP assays.
More description
|
|
| DC21264 | M4284 |
M4284 (M-4284) is a high-affinity biphenyl mannoside FimH inhibitor with HAI of 16 nM in hemagglutination assay.
More description
|
|
| DC23382 | L-α-Hydroxyglutaric acid disodium salt |
L-α-Hydroxyglutaric acid disodium salt (L-2-Hydroxyglutarate.
More description
|
|
| DC21262 | LYS228 |
LYS228 is a novel potent monobactam that shows potent activity against carbapenem-resistant isolates of Enterobacteriaceae with MIC90 of 2 ug/ml.
More description
|
|
| DC20440 | LYPLAL1-IN-11 |
LYPLAL1-IN-11 is a potent, selective, covalent inhibitor of lysophospholipase-like 1 (LYPLAL1) with IC50 of 6 nM.
More description
|
|
| DC21260 | LY-83583 |
LY-83583 is an inhibitor of soluble guanylate cyclase (sGC) and of cGMP production, inhibits sGC in human platelets with IC50 of 2 uM.
More description
|
|
| DC23636 | LY-503430 |
LY-503430 is a novel AMPA receptor (AMPAR) potentiator that selectively enhances glutamate-induced calcium influx into HEK293 cells transfected with human GLUA1, GLUA2, GLUA3, or GLUA4 AMPA receptors with EC50 of 0.1-4 uM.
More description
|
|
| DC7459 | LY500307 |
LY500307 is a potent, selective estrogen receptor β agonist with EC50 of 0.66 nM, 32-fold selectivity against estrogen receptor α. Phase 2.
More description
|
|
| DC22420 | LY-487379 hydrochloride |
LY-487379 is a potent, selective positive allosteric modulator of mGluR2 (EC50=0.27 uM) without activity at mGluR3.
More description
|
|
| DC23597 | LY-404187 |
LY-404187 is a potent, selective, positive allosteric modulator of AMPA receptor (AMPAR) with EC50 of 1.3 uM.
More description
|
|
| DC23829 | LY379196 |
LY379196 is a highly specific PKCβ inhibitor with IC50 of 30-50 nM for PKCβI and PKCβII, displays >10-fold selectivity of PKCα, δ, η, and γ (>600 nM).
More description
|
|
| DC11323 | LY354740 |
LY354740 is an agonist of the group II metabotropic glutamate receptor (mGluR) subtypes mGluR2 and mGluR3 (Kis = 99 and 94 nM, respectively).
More description
|
|
| DC9314 | LY 344864 |
LY344864 is a selective receptor agonist with an affinity of 6 nM (Ki) at the recently cloned 5-HT1F receptor.
More description
|
|