| DC10448 |
Belizatinib(TSR-011)
Featured
|
Belizatinib(TSR-011) is an orally available inhibitor of both the receptor tyrosine kinase anaplastic lymphoma kinase (ALK) and the tropomyosin-related kinases (TRK) TRKA, TRKB, and TRKC, with potential antineoplastic activity. |

|
| DC9956 |
BFH772
Featured
|
BFH772 is a potent oral VEGFR2 inhibitor, which is highly effective at targeting VEGFR2 kinase with an IC50 value of 3 nM. |

|
| DC10335 |
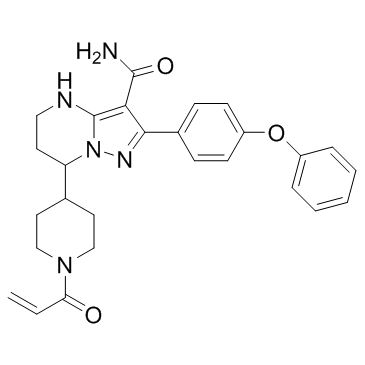
BGB-3111 |
BGB-3111 is a potent, selective and orally available Bruton's tyrosine kinase (Btk) inhibitor. |
|
| DC5194 |
NVP-BGJ398(Infigratinib)
Featured
|
BGJ398 (NVP-BGJ398) is a potent and selective FGFR inhibitor for FGFR1/2/3 with IC50 of 0.9 nM/1.4 nM/1 nM, >40-fold selective for FGFR versus FGFR4 and VEGFR2, and little activity to Abl, Fyn, Kit, Lck, Lyn and Yes. Phase 2. |

|
| DC11760 |
BI 885578 |
BI 885578 (BI-885578, BI885578) is a potent,selective, ATP-competitive IGF1R/InsR tyrosine kinase inhibitor with Kd of 9 nM/12 nM, IC50 of 1 nM/1 nM, respectively. |

|
| DC10530 |
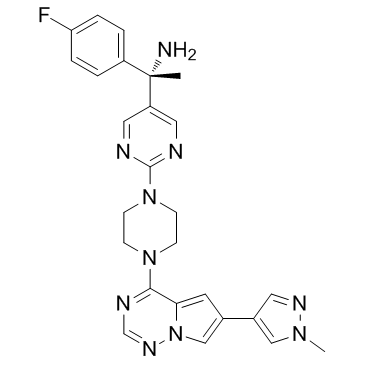
BLU-285 (Avapritinib)
Featured
|
BLU-285 is a potent and selective exon 17 mutant KIT kinase inhibitor with IC50 of 0.27 nM for KIT D816V. |
|
| DC10092 |
BLU554(Fisogatinib)
Featured
|
BLU-554 is a potent fibroblast growth factor receptor 4 (FGFR4) inhibitor. |

|
| DC11479 |
BLU-667 (Pralsetinib)
Featured
|
BLU-667 (Pralsetinib) is a highly potent, selective, next generation RET inhibitor with IC50 of 0.3-0.4 nM for WT RET, RET mutants V804L, V804M, M918T and CCDC6-RET fusion. |

|
| DC8280 |
BLU-9931
Featured
|
BLU9931 is the first selective small molecule inhibitor of FGFR4 with IC50 of 3 nM; less potent for FGFR1/2/3(IC50> 150 nM). |

|
| DC8103 |
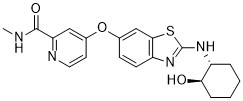
BLZ-945
Featured
|
BLZ945 is a highly selective and brain-penetrant inhibitor of CSF1R with IC50 of 1 nM; >3200-fold higher than its affinity for other kinases. |
|
| DC8515 |
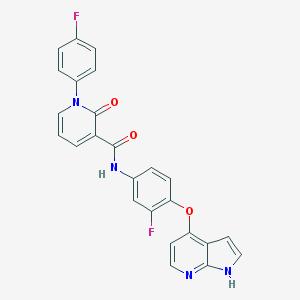
BMS-2 |
BMS-2 is a potent MET kinase inhibitor with an IC50 value of 1.8nM. It also possesses potent inhibitory activity against Flt-3 and VEGFR-2 kinases, IC50 values of 4nM and 27nM respectively. |
|
| DC5037 |
BMS-536924
Featured
|
BMS-536924 is an ATP-competitive IGF-1R inhibitor with IC50 of 100 nM, modest activity for Mek, Fak, and Lck with very little activity for Akt1, MAPK1/2. |

|
| DC3105 |
BMS-599626 (AC480)
Featured
|
BMS-599626 (AC480) is a selective and efficacious inhibitor of HER1 and HER2 with IC50 of 20 nM and 30 nM, respectively. |
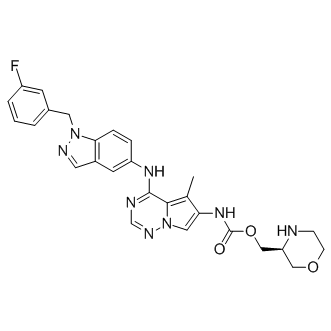
|
| DC3126 |
BMS754807
Featured
|
BMS-754807 is a potent and reversible inhibitor of IGF-1R/IR family kinases, inhibits IGF-1R, IR, Met, TrkA and TrkB with IC50 of 1.8 nM, 1.7 nM, 5.6 nM, 7.4 nM and 4.1 nM, respectively. |

|
| DC9638 |
BMS-794833 |
BMS-794833 is a potent ATP competitive inhibitor of Met/VEGFR2 with IC50 of 1.7/15 nM; also inhibits Ron, Axl and Flt3 with IC50 of <3 nM; a prodrug of BMS-817378.
|
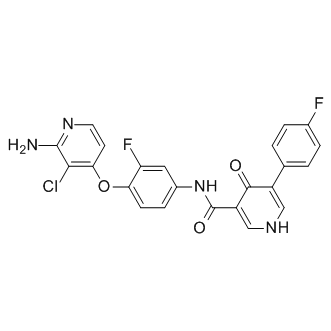
|
| DC11819 |
BMS-986142
Featured
|
BMS-986142 is a potent, selective, and reversible BTK (Bruton’s tyrosine kinase) inhibitor (BTK IC₅₀ = 0.5nM; human WB IC₅₀ = 90 nM). |

|
| DC12030 |
BMS-986195
Featured
|
BMS-986195 is a potent, covalent, irreversible inhibitor of Bruton’s tyrosine kinase (BTK). |
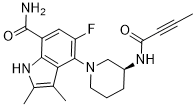
|
| DC2019 |
Brivanib (bms-540215)
Featured
|
Brivanib is an ATP-competitive inhibitor against human VEGFR2 and FGFR with IC50 of 25 nM and 148 nM, respectively. |

|
| DC9266 |
Cabozantinib S-malate
Featured
|
Cabozantinib S-malate (XL184, BMS-907351) is a potent VEGFR2 inhibitor with IC50 of 0.035 nM and also inhibits c-Met, Ret, Kit, Flt-1/3/4, Tie2, and AXL with IC50 of 1.3 nM, 4 nM, 4.6 nM, 12 nM/11.3 nM/6 nM, 14.3 nM and 7 nM, respectively. |

|
| DC3138 |
Canertinib dihydrochloride
Featured
|
Canertinib dihydrochloride is the hydrochloride salt of an orally bio-available quinazoline with potential antineoplastic and radiosensitizing activities. |

|
| DC8482 |
CAY10415(MSDC-0160)
Featured
|
CAY10415 is a potent, antidiabetic drug of the TZD structural class. |

|
| DC3170 |
Cediranib
Featured
|
Cediranib (AZD2171) is a highly potent VEGFR2 inhibitor with IC50 of 0.5 nM, also inhibits Flt1/4 with IC50 of 5 nM/≤3 nM. |
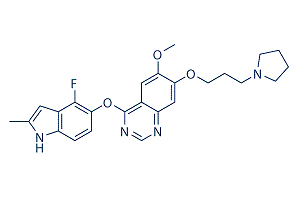
|
| DC8138 |
CEP-28122
Featured
|
CEP-28122 is a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. |

|
| DC8009 |
CEP-37440
Featured
|
CEP-37440 is a novel potent and selective Dual FAK/ALK inhibitor with IC50 s of 2.3 nM (FAK) and 120 nM(ALK cellular IC50 in 75% human plasma). |

|
| DC10337 |
CEP-40783
Featured
|
CEP-40783 is a potent, selective and orally available inhibitor of AXL and c-Met with IC50 values of 7 nM and 12 nM, respectively. |
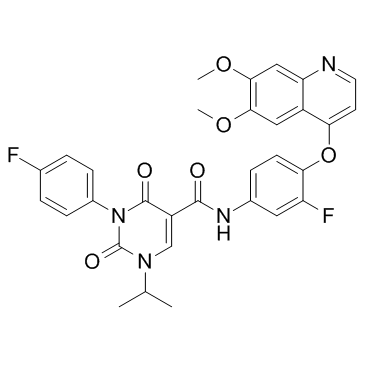
|
| DC7380 |
CGI-1746
Featured
|
CGI1746 is a small-molecule Btk inhibitor chemotype with a new binding mode that stabilizes an inactive nonphosphorylated enzyme conformation. |

|
| DC8418 |
CH5183284 (Debio-1347)
Featured
|
CH5183284 is a selective and orally available FGFR inhibitor with IC50 of 9.3 nM, 7.6 nM, 22 nM, and 290 nM for FGFR1, FGFR2, FGFR3, and FGFR4, respectively. Phase 1. |
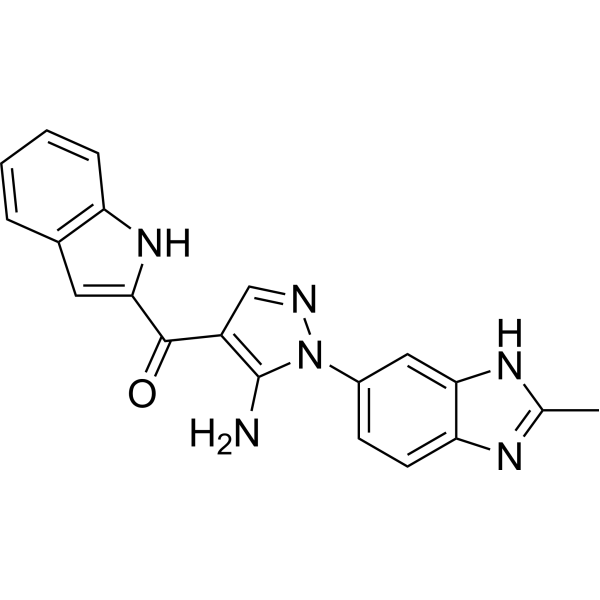
|
| DC7383 |
CH5424802(Alectinib)
Featured
|
CH5424802(AF 802; Alectinib) is a potent ALK inhibitor with IC50 of 1.9 nM, sensitive to L1196M mutation. |
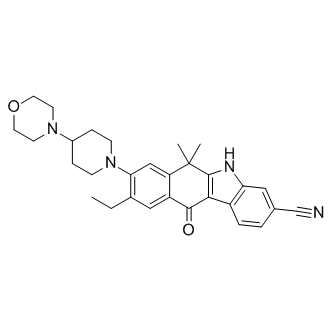
|
| DC8728 |
CH5424802(Alectinib HCl)
Featured
|
CH5424802(AF 802; Alectinib) is a potent ALK inhibitor with IC50 of 1.9 nM, sensitive to L1196M mutation. |

|
| DC10467 |
CHMFL-BMX-078
Featured
|
CHMFL-BMX-078 is a highly potent and selective type II irreversible BMX kinase inhibitor with an IC50 of 11 nM. |

|
 DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.
DC Chemicals' products qualify for U.S. tariff exemptions. We guarantee no price increases due to customs duties and maintain stable supply, continuing to deliver reliable research solutions to our American clients.