 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC7045 | A-769662 Featured |
A-769662 is a potent, reversible AMPK activator with EC50 of 0.8 μM, little effect on GPPase/FBPase activity.
More description
|

|
| DC5163 | A-674563 Featured |
A-674563 is a potent, orally available Akt1 inhibitor with an IC50 value of 11nM. Also displays activity against PKA and CDK2 with IC50 values of 16nM and 46nM respectively.
More description
|
|
| DC8406 | A-438079 HCl Featured |
A-438079 HCl is a potent, and selective P2X7 receptor antagonist with pIC50 of 6.9.
More description
|

|
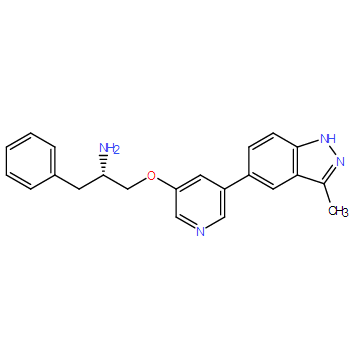
| DC9574 | A-317491 (sodium salt hydrate) Featured |
A-317491 is a non-nucleotide P2X3 and P2X2/3 receptor antagonist, which inhibits calcium flux mediated by the receptors.
More description
|
|
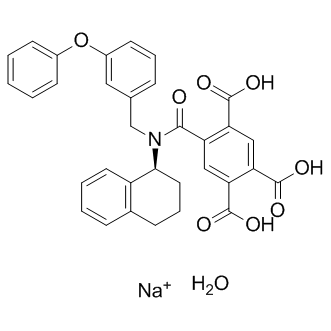
| DC12702 | A1874 Featured |
A1874 (A-1874) is a nutlin-based and BRD4-degrading PROTAC with DC50 of 32 nM (induce BRD4 degradation in cells).
More description
|
|
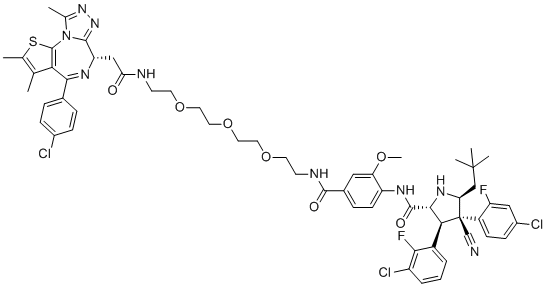
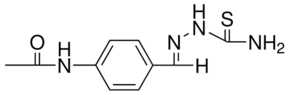
| DC7517 | THIACETAZONE Featured |
A thiosemicarbazone that is used in association with other antimycobacterial agents in the initial and continuation phases of antituberculosis regimens. Thiacetazone containing regimens are less effective than the short-course regimen recommended by the I
More description
|
|
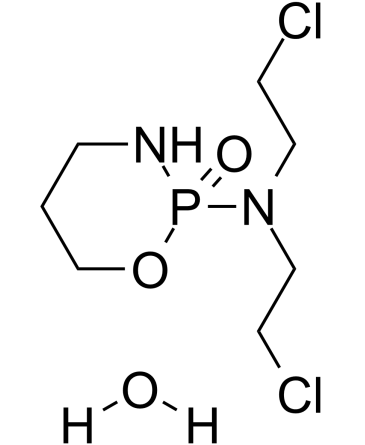
| DC24191 | Cyclophosphamide hydrate Featured |
A synthetic alkylating agent chemically related to the nitrogen mustards with antineoplastic and immunosuppressive activities.
More description
|
|
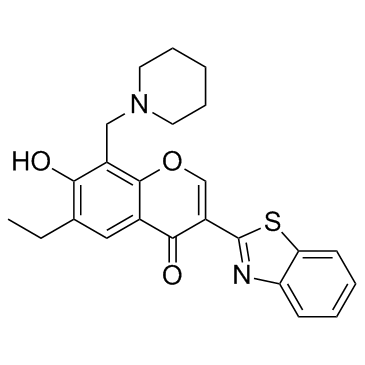
| DC23210 | SZL P1-41 Featured |
A small molecule Skp2 E3 ligase inhibitor that prevents Skp2-Skp1 interaction and Skp2 SCF E3 ligase activity in vitro.
More description
|
|
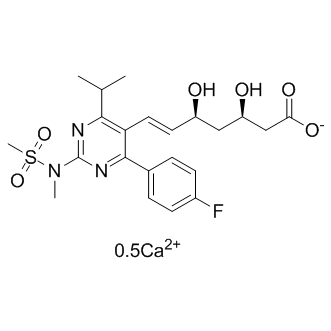
| DCAPI1468 | Rosuvastatin Calcium Featured |
A selective, competitive inhibitor of HMG-CoA reductase, that is also antilipemic.
More description
|
|
| DC7029 | SC-26196 Featured |
A selective Δ6 desaturase inhibitor that displays selectivity over Δ5 and Δ9 desaturases
More description
|

|
| DC7054 | AM580 Featured |
A retinoic acid analog and selective RARα agonist
More description
|

|
| DC23938 | Esomeprazole sodium Featured |
A proton pump inhibitor that reduces stomach acid secretion by inhibition of the H+/K+-ATPase in the parietal cells.
More description
|

|
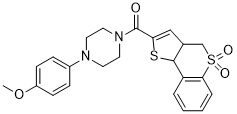
| DC21321 | ML349 Featured |
A potent, specific acyl protein thioesterase 2 (APT-2, LYPLA2) inhibitor with IC50 of 144 nM and Ki of 120 nM, >20-fold selectivity over APT-1 and serine hydrolase enzyme family.
More description
|
|
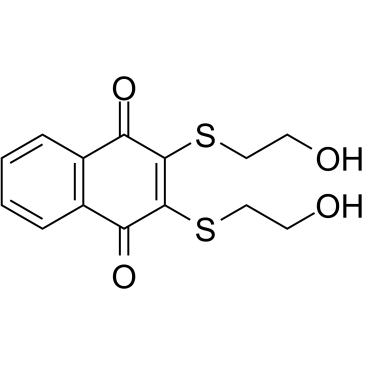
| DC11691 | NSC95397 Featured |
A potent, selective, reversible Cdc25 dual specificity phosphatase inhibitor with Ki of 32/96/40 nM for Cdc25A/B/C, respectively.
More description
|
|
| DC22536 | CFMTI Featured |
A potent, selective, allosteric and oral mGluR1 antaognist with IC50 of 2.6 and 2.3 nM for human and rat mGluR1, respectively.
More description
|

|
| DC22625 | Rolipram Featured |
A potent, selective inhibitor of phosphodiesterases PDE4 with IC50 of 3 nM, 130 nM and 240 nM for PDE4A, PDE4B, and PDE4D, respectively.
More description
|

|
| DC21288 | MK-8617 Featured |
A potent, orally active pan-inhibitor of HIF prolyl hydroxylase (HIF-PHD) with IC50 of 1.0,1.0, and 14 nM for HIF-PHD1, 2, and 3, respectively.
More description
|

|
| DC11641 | Cridanimod Featured |
A potent type I interferon (IFN) inducer that directly binds to STING and triggers a strong antiviral response through the TBK1/IRF3 route.
More description
|

|
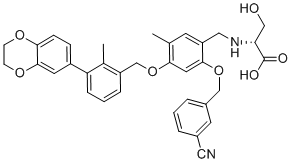
| DC22928 | BMS-1001 Featured |
A potent PD-1/PD-L1 interaction inhibitor with IC50 of 2.25 nM in a homogenous time-resolved fluorescence binding assay..
More description
|
|
| DC22340 | Linaclotide Featured |
A potent and selective GC-C agonist (Ki=1.23-1.64 nM) that elicits pharmacological effects locally in the gastrointestinal tract.
More description
|

|
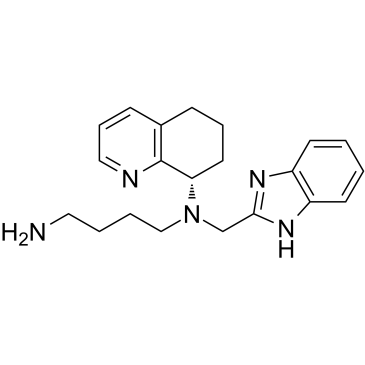
| DC22584 | Mavorixafor(AMD-070) Featured |
A potent and selective antagonist of CXCR4 with IC50 of 13 nM in a CXCR4 125I-SDF inhibition binding assay.
More description
|
|
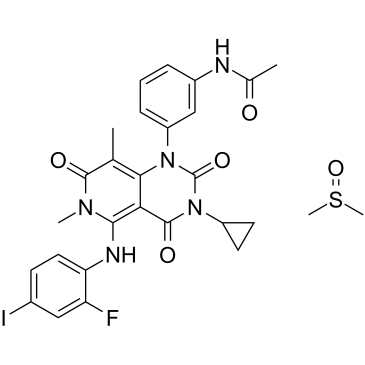
| DC22627 | Trametinib DMSO solvate Featured |
A potent and highly specific MEK1/2 inhibitor with IC50 of 0.92 nM/1.8 nM.
More description
|
|
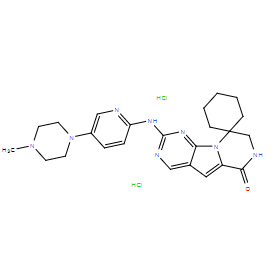
| DC21033 | Trilaciclib hydrochloride(G1T28) Featured |
A novel, potent and selective inhibitor of CDK4/6 with biochemical IC50 of 1 nM and 4 nM for CDK4/cyclin D1 and CDK6/cyclin D3, respectively.
More description
|
|
| DC5893 | SCD1 inhibitor Featured |
A novel stearoyl-CoA desaturase1 (SCD1) inhibitor.The compound exhibited robust in vivo activity with dose-dependent desaturation index lowering effects.
More description
|

|
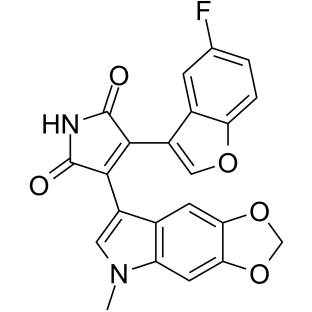
| DC11827 | 9-ING-41 Featured |
9-ING-41 is a potent glycogen synthase kinase-3 (GSK-3) inhibitor[1]. 9-ING-41 induces apoptosis and cell cycle arrest at prophase by targeting centrosomes and microtubule-bound GSK-3β. 9-ING-41 has anticancer activity[2].
More description
|
|
| DC23203 | E4CPG Featured |
A non-selective group I/group II metabotropic glutamate receptor (mGluR) antagonist..
More description
|

|
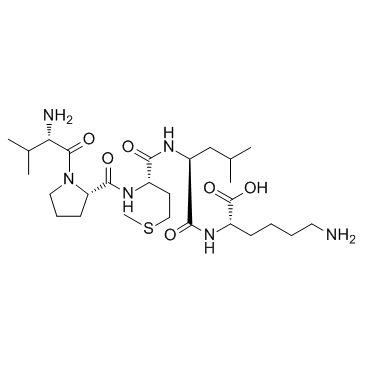
| DC23206 | Bax inhibitor peptide V5 Featured |
A cell-permeable synthetic peptide inhibitor of Bax conformational change and mitochondrial translocation.
More description
|
|
| DC9628 | A 419259 (trihydrochloride) Featured |
A 419259 3Hcl is an apoptosis inducing agent that inhibits Src family kinases (c-Src).
More description
|

|
| DC8388 | 8-Bromo-cAMP Featured |
8-Bromo-cAMP is a cell perbeable cyclic AMP (cAMP) analog and a PKA activator.
More description
|

|
| DC7876 | 7ACC2 Featured |
7ACC2 is a new potent MCT inhibitor with IC50 of 11 nM for inhibition of [14C]-lactate influx; new antitumor treatment targeting lactate transport in cancer cells.
More description
|

|