 To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
To enhance service speed and avoid tariff delays, we've opened a US warehouse. All US orders ship directly from our US facility.
| Cat. No. | Product Name | Field of Application | Chemical Structure |
|---|---|---|---|
| DC26053 | Benzamil (hydrochloride) Featured |
Benzamil hydrochloride is a specific and potent sodium channel (ENaC) blocker.
More description
|

|
| DC67639 | N-Α-BENZOYL-L-ARGININE ETHYL ESTER HYDROCHLORIDE Featured |

|
|
| DC67638 | N-Benzoyl-L-arginine ethyl ester hydrochloride Featured |
N-Benzoyl-L-arginine ethyl ester hydrochloride is an arginine derivative.
More description
|

|
| DC7816 | MG-132 Featured |
MG-132 is an inhibitor of proteasome with IC50 of 100 nM, and also inhibits calpain with IC50 of 1.2 μM.
More description
|

|
| DC32972 | H2DCFDA Featured |
H2DCFDA is a fluorescent cell permeabl ROS indicator.
More description
|

|
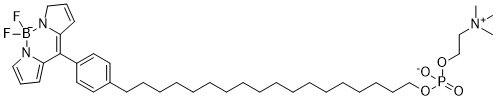
| DC9797 | CLR1501 Featured |
CLR1501 is a synthetic analogs of the tumor-targeting agent alkylphosphocholine (APC), which is specifically attracted to cancer cells.
More description
|
|
| DC9803 | CMP8 Featured |
CMP8 is a high-affinity ligand that binds only to mutant ERLBD.
More description
|

|
| DC40144 | TTA-A2 Featured |
TTA-A2 is a potent, selective and orally active t-type voltage gated calcium?channel antagonist with reduced pregnane X receptor (PXR) activation. TTA-A2 is equally potent against the Cav3.1 (a1G) and Cav3.2 (a1H) channels with IC50 values of 89 nM and 92 nM, respectively, at -80 and -100 mV holding potentials. TTA-A2 can be used for the research of a variety of human neurological diseases, including sleep disorders and epilepsy.
More description
|

|
| DC67637 | (1R)-1-[5-(2,2,2-trifluoroethoxy)-2-pyridyl]ethanamine Featured |
-1-[5-(2,2,2-trifluoroethoxy)-2-pyridyl]ethanamine.gif)
|
|
| DC67636 | 2-(4-Cyclopropylphenyl)acetic acid Featured |

|
|
| DC67635 | (S)-GSK-F1 Featured |
(S)-GSK-F1 (Compound 28) is an inhibitor for type III phosphatidylinositol 4-kinase α (PI4KIIIα). (S)-GSK-F1 inhibits PI4Kα, PI4Kβ, PI4Kγ, PI3Kα, PI3Kβ and PI3Kδ with pIC50 of 8.3, 6.0, 5.6, 5.6, 5.1 and 5.6, respectively. (S)-GSK-F1 exhibits anti-hepatitis C virus (HCV) activity through inhibition of HCV replication. (S)-GSK-F1 exhibits moderate pharmacokinetic characters in rat model.
More description
|
-GSK-F1.gif)
|
| DC67634 | 2,5-dimethylthiophen-3-ylboronic acid Featured |

|
|
| DC28976 | Dizocilpine free base Featured |
Dizocilpine, a potent anticonvulsant, is a selective and non-competitive NMDA receptor antagonist, with a Kd of 37.2 nM in rat brain membranes. Dizocilpine acts by binding to a site located within the NMDA associated ion channel and thus prevents Ca2+ flux.
More description
|

|
| DC4128 | Ivacaftor (VX-770) Featured |
Ivacaftor (VX-770, Kalydeco) is a potentiator of CFTR targeting G551D-CFTR and F508del-CFTR with EC50 of 100 nM and 25 nM, respectively.
More description
|

|
| DC77410 | BMS-986449 Featured |
BMS-986449 is a CELMoD molecular glue and an IKZF2/IKZF4 degrader. BMS-986449 targets the degradation of transcription factors Helios (IKZF2) and Eos (IKZF4) in regulatory T (Treg) cells. BMS-986449 redirects the E3 ubiquitin ligase Cereblon to induce the degradation of Helios and Eos, reprogramming Treg cells and enhancing antitumor immunity. BMS-986449 is promising for research of advanced solid tumors.
More description
|

|
| DC10670 | CNQX Featured |
CNQX is a potent AMPA/kainate receptor antagonist. Also antagonist at glycine modulatory site on NMDA receptor complex.
More description
|

|
| DC9341 | Naspm Featured |
Naspm(1-Naphthylacetyl spermine) is a potent and selective Ca2+ permeable AMPA receptor(Ca-perm AMPAR) blocker.
IC50 value:
Target: CP-AMPAR blocker
Naspm is a selective blocker of Ca(2+)-permeable GluA2-lacking AMPA receptors, reduced MNNG-induced CA1
More description
|

|
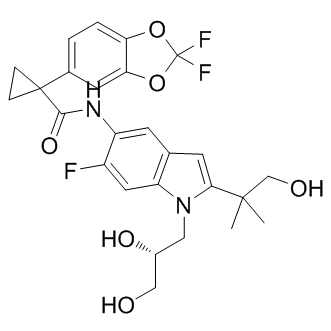
| DC7531 | Tezacaftor(VX661) Featured |
Tezacaftor(VX661) is a second F508del CFTR corrector and is believed to help CFTR protein reach the cell surface.
More description
|
|
| DC24126 | Triptorelin Featured |
Triptorelin is a synthetic decapeptide agonist analog of luteinizing hormone releasing hormone (LHRH), shows greater potency than endogenous LHRH, triptorelin reversibly represses gonadotropin secretion..
More description
|

|
| DC10238 | Fluoxymesterone Featured |
Fluoxymesterone is an Androgen. The mechanism of action of fluoxymesterone is as an Androgen Receptor Agonist.
More description
|

|
| DC67631 | BHEM-Cholesterol, Iodide Featured |

|
|
| DC67630 | BHEM-Cholesterol, bromide Featured |

|
|
| DC67629 | BHE-Cholesterol Featured |

|
|
| DCG-002 | β-Sitosterol Featured |
β-Sitosterol is orally active. Beta-Sitosterol exhibits multiple activities, including anti-inflammatory, anticancer, antioxidant, antimicrobial, antidiabetic, antioxidant enzyme, and analgesic. Beta-Sitosterol inhibits inflammation and impaired adipogenesis in bovine mammary epithelial cells by reducing levels of ROS, TNF-α, IL-1β, and NF-κB p65 and restoring the activity of the HIF-1α/mTOR signaling pathway. Beta-Sitosterol induces apoptosis in cancer cells through ROS-mediated mitochondrial dysregulation and p53 activation. Beta-Sitosterol exerts its anticancer effects in cancer cells by activating caspase-3, caspase-8, and caspase-9, mediating PARP inactivation, MMP loss, altered Bcl-2-Bax ratio, and cytochrome c release. Beta-Sitosterol modulates macrophage polarization and reduces rheumatoid inflammation in mice. Beta-Sitosterol inhibits tumor growth in multiple mouse cancer models. Beta-Sitosterol can be used in the research of arthritis, lung cancer, breast cancer and other cancers, diabetes, etc.
More description
|

|
| DCD-042 | Stigmasterol Featured |
Stigmasterol is an orally acitve, immunomodulatory agent with anti-inflammatory and neuroprotective effect, as well as able to cross the blood-brain barrier. Stigmasterol activates AMPK, which in turn inhibits NF-κB and NLRP3 signaling pathways, reduces microglia-mediated neuroinflammation, and alleviates cognitive impairment and Alzheimer's disease. Stigmasterol regulates M1/M2 polarization of microglia through the TLR4/ NF-κB pathway, thereby reducing neuropathic pain. Stigmasterol can be used for neurodegenerative diseases, inflammatory diseases, and pain management, among others.
More description
|

|
| DC36834 | Sitostanol Featured |
Sitostanol is a saturated sterol found in a variety of plants, including corn, wheat, and rice and reduces cholesterol uptake in rats and rabbits.
More description
|

|
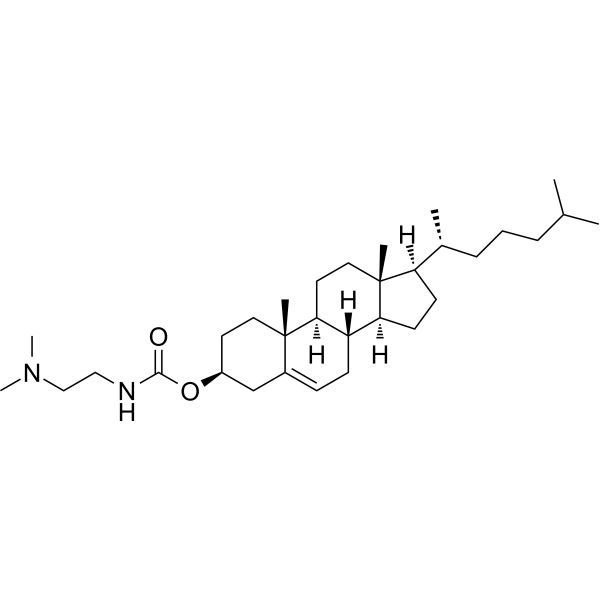
| DC66108 | DC-cholesterol Featured |
DC-cholesterol(3β-[N-(N′,N′-Dimethylaminoethyl)carbamoyl]cholesterol), a lipid, has been investigated in cancer gene therapy and vaccine delivery system.
More description
|
|
| DC67627 | CHEMS-PEG-5000 Featured |

|
|
| DC67626 | CHEMS-PEG-2000 Featured |

|
|
| DC67624 | SCS Featured |
SCS is an orally active cholesterol biosynthesis agonist that targets NPC2. It activates SREBP2 by competitively binding to NPC2, thereby promoting cholesterol synthesis (EC50 = 50 μM).
More description
|

|